做功能基因定位的生物狗基本都知道,经过10来年的发展,关联分析与连锁分析一样,已经成为一项基本工具,广泛应用于动植物功能基因挖掘中。在动植物(尤其是植物)的关联分析中,TASSEL软件是最早出现的开源软件,相对于其他软件,TASSEL也是使用的最广泛的,您想不想知道在您有了表型、基因型之后,怎么使用TASSEL进行关联分析呢?往下看吧

,小编教你啊~

先给软件的下载地址 http://www.maizegenetics.net/tassel
在拿到表型和基因型(我们以vcf为例)后,完成GWAS分析,你只需要5步。
第一步 数据质控
内容包括:
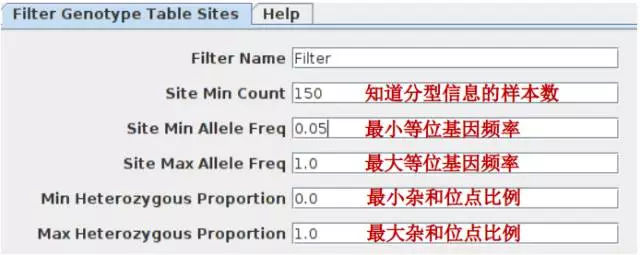
1)按分型百分比条件过滤,多数文章剔除缺失率在20%以上的位点,样本量较大的群体中,可以将缺失率小于50%的位点都保留;
2)按等位基因频率过滤,通常去除最小(或第二)等位基因频率小于5%的位点,样本量较大的群体中,可以降低到1%;
3)多等位位点的过滤(当软件无法接受时);
4)有时候还会去除缺失数据太多的样本(基因型缺失比例大于20%或更高);
5)哈迪温伯格平衡过滤,一般在无法使用较为复杂的统计模型的情况下使用,如人类的Case/Control GWAS中一般将不符合哈迪温伯格平衡的位点过滤掉,动植物GWAS中一般不过滤;
6)表型极端值去除,用Excel从小到大排一下序看看就知道了。
如果你熟悉plink软件(https://www.cog-genomics.org/plink2),那么第1)到第5)点就变得非常easy了,一条命令行即可以搞定:
./plink --vcf ./test.vcf --maf 0.05 --geno 0.2 --mind 0.2 --hwe 0.001 --biallelic-only --recode vcf-iid --out test.bia.maf0.05.int0.8.ind0.8.hwe0.001 --allow-extra-chr
--vcf 表示输入的文件为vcf文件
--maf 控制第二等位基因频率的,我们这里设置为不小于5%
--geno 控制位点基因型的缺失比例的,我们这里设置为20%,即缺失比例大于20%的位点都会过滤掉
--mind 控制样本基因型缺失比例的,我们这里设置为20%,即基因型缺失比例大于20%的位点都会被过滤掉
--hwe 哈迪温伯格平衡显著性阈值的,我们这里设置为0.001,即哈迪温伯格平衡检验p值小于0.001的位点都会被过滤掉
--biallelic-only 表示只保留二等位位点
--recode 把基因编码为某种格式,我们这里还是输出为与输入文件一样的vcf,所以选vcf-iid,另外还有十几种格式,有需要的生物狗狗们可以到plink的官网上看看
--out 给定输出文件的前缀
--allow-extra-chr 允许存在22+X+Y以外的染色体,非人类数据建议都把这一项给上
我们要讲的TASSEL软件,也可以完成一些过滤,导入数据vcf数据后(File -- open),选中导入的基因型文件,点击Filter菜单操作就行了,具体如下图。
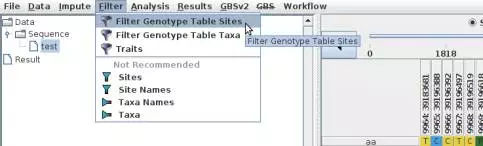
第二步 群体结构分析
群体结构分析的内容其实是很丰富的,包括系统发育树的构建(用于直观地看出样本的亚群分化情况);基于模型的(model-base)群体结构分析,可以使用的软件包括STRUCTURE、Admixture等,分析结果可以获得Q矩阵,用于关联分析;数学降维的主成分分析,也就是我们常说的PCA分析,获得样本的主成分得分表,也可以作为Q矩阵用于关联分析。
本帖小编带领大家用TASSEL进行PCA分析,获得的结果作为Q矩阵用于关联分析,系统发育树的构建(各种树)方法可以参考百迈客云课堂(http://live.biocloud.net/course/21)相关的课程,STRUCTURE的使用介绍见今天下午的直播课程或者云课堂。
PCA分析:导入vcf后,选中导入的基因型文件,依次选择Analysis--Relatedness--PCA即可。
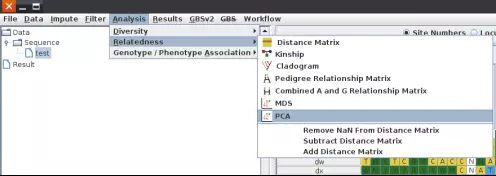
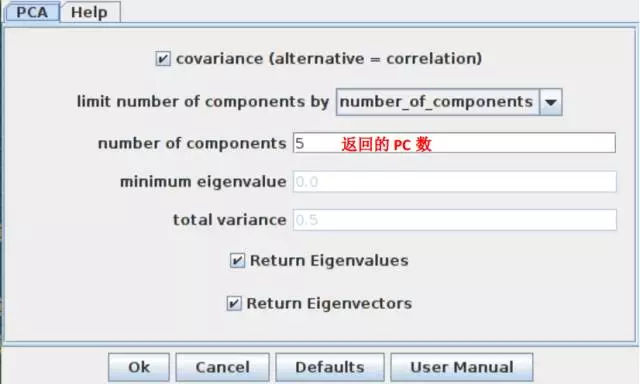
获得的结果包括样本在各个PC(上面选返回多少个就显示多少个的信息)的得分表以及每个PC的特征值列表;如下两图:
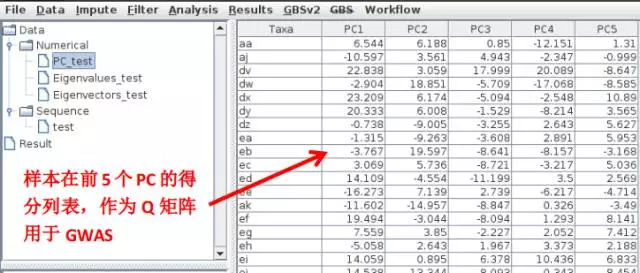
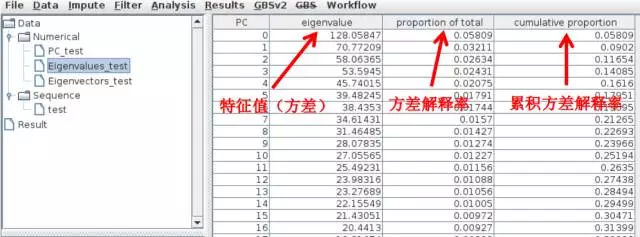
第三步 亲缘关系分析
亲缘关系衡量的是两两样本间的相关关系(可能是血缘的或者其他的)的数值,计算的方法很多(具体介绍见云课堂GWAS生信培训班),亲缘关系矩阵(K)在MLM模型中作为随机效控制关联结果的假阳性。在TASSEL中,基于分子标记,获得亲缘关系矩阵有两种方法,一种是计算Distance Matrix(Analysis -- Distance Matrix),另一种是计算Kinship(Analysis -- Kinship),两种途径获得的结果都可以用于关联分析,没有说非要用哪一种才是最好的。
我们以计算Kinship为例,操作图示如下,OK以后即可得到结果。
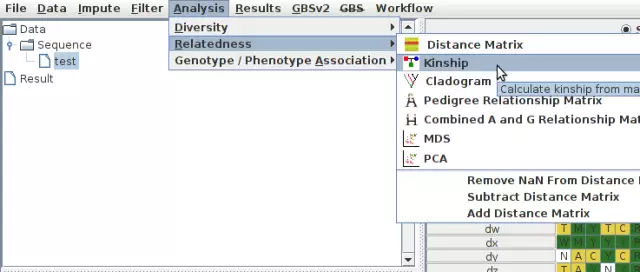
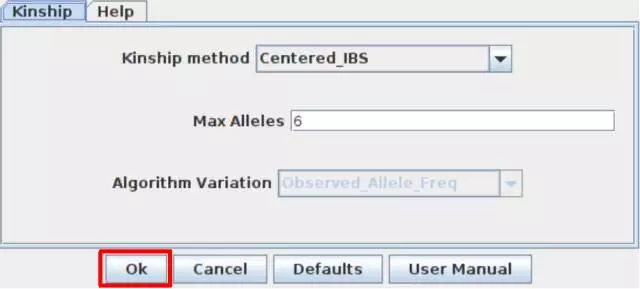
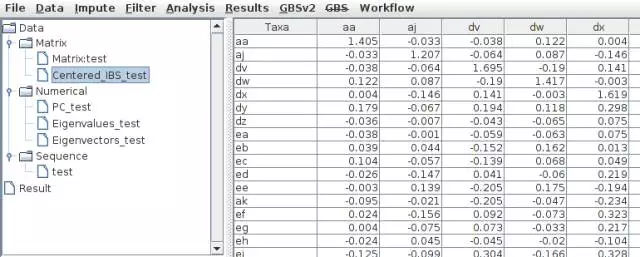
第四步 关联分析
获得Q矩阵,K矩阵后,导入性状数据,我们就可以进行关联分析了,关联分析之前,我们需要将基因型、性状、Q矩阵合并:按Ctrl键,鼠标依次选择基因型列表、样本在前5个PC的得分矩阵、性状列表,选择Data--Intersect jion完成合并。
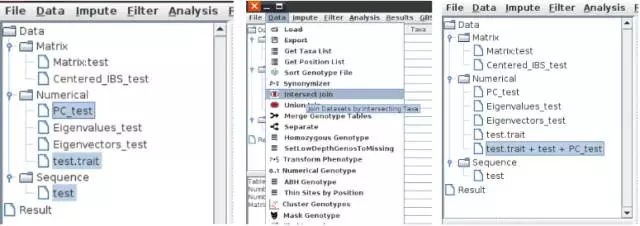
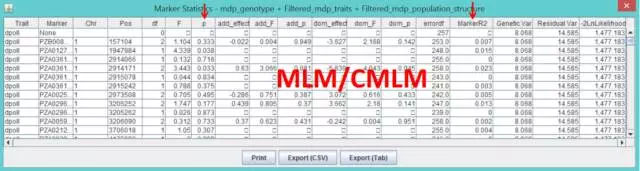
终于进入正题了,下面的操作将教会大家如何用TASSEL完成GLM和MLM/CMLM的关联分析。
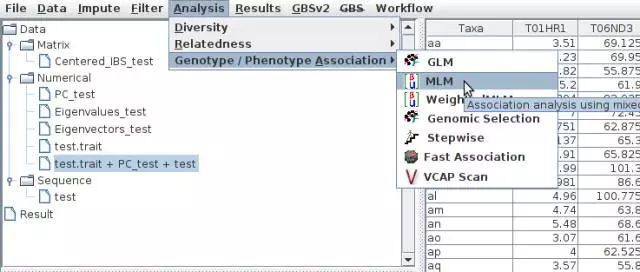
GLM:选中性状+基因型+PC列表;Analysis--Genotype/Phenotype Association--GLM;选择输出路径,填写输出文件名,然后点击OK即可。
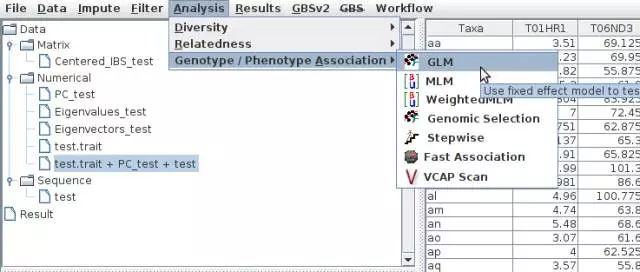
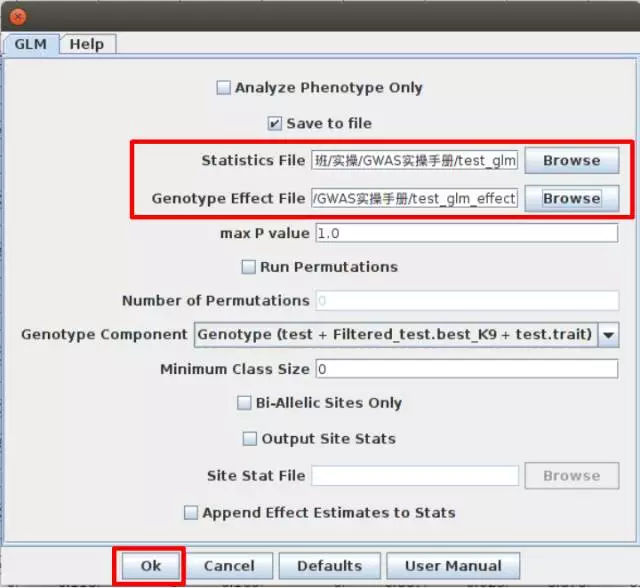
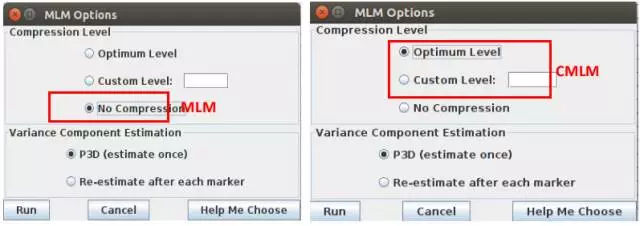
MLM/CMLM:选中性状+基因型+PC列表和亲缘关系列表,Analysis--Genotype/Phenotype Association--GLM;选择输出路径,填写输出文件名,然后点击Okay即可。
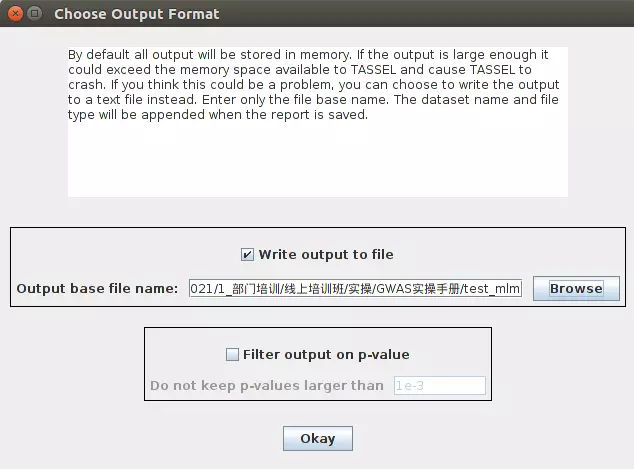
结果如下
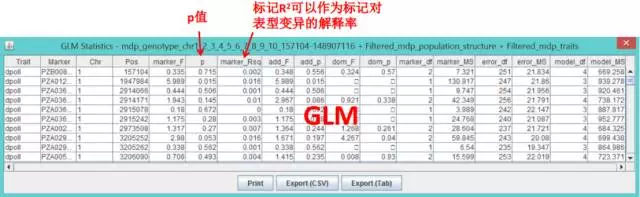
第五步 绘图展示
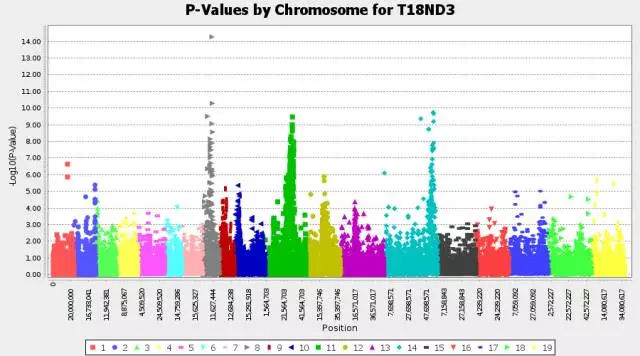
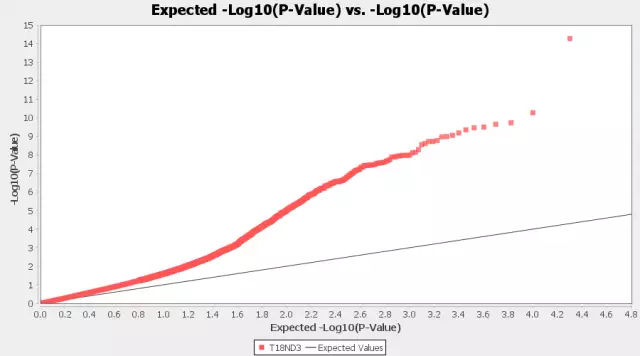
把结果文件导入TASSEL中,选中导入的文件,Results--Manhattan plo/QQ plot即可绘出相关图形(TASSEL自带绘图功能画出来的图,小编真心觉得挺丑);想知道漂亮的图怎么画出来的吗,请参加今天下午的直播课程吧,小编手把手教你,包教包会。
通过上面的学习,相信大家已经学会了都动植物经典关联分析软件TASSEL的使用,以上展示的是该软件界面版的使用方法,界面版的软件在数据量比较小的时候操作起来比较顺畅,但是,如果数据量比较大的话,界面版的可能hold不住了,这时您可能想起要用命令行版的,那命令行版该怎么使用呢,请参加下午四点半的直播培训,我们一一为您讲解。
TASSEL目前只能实现GLM/MLM/CMLM三种模型,其他常用模型如EMMAX、FaST-LMM(样本间亲缘关系对结果的影响比较大的时候建议选择这两种模型)等的使用方法以及GAPIT、Admixture软件的使用方法,请看百迈客云课堂GWAS生信专题培训班的内容(http://live.biocloud.net/course/21)。

想要参加今天下午的直播培训的老师们,请下载演示数据,链接: https://pan.baidu.com/s/1nv9TFM5 密码: up8d;并提前配置好Java环境,安装好以下软件:
STRUCTURE:https://web.stanford.edu/group/pritchardlab/structure.html
CLUMMP:https://rosenberglab.stanford.edu/clumpp.html
R:https://www.r-project.org/
Pophelper:http://royfrancis.github.io/pophelper/
CMplot:https://github.com/YinLiLin/R-CMplot
转自:百迈克
- 本文固定链接: https://maimengkong.com/zu/1454.html
- 转载请注明: : 萌小白 2023年4月16日 于 卖萌控的博客 发表
- 百度已收录
