本篇文章给大家分享的是有关怎么使用ROSE鉴定超级增强子,小编觉得挺实用的,因此分享给大家学习,希望大家阅读完这篇文章后可以有所收获,话不多说,跟着小编一起来看看吧。
ROSE是最经典的超级增强子预测软件,由Richard A. Young大牛团队开发,源代码的网址如下
http://younglab.wi.mit.edu/super_enhancer_code.html
首先通过Oct4, Sox2, Nanog这3种转录因子的chip数据去识别小鼠胚胎干细胞中的增强子区域,鉴定到了8794个增强子区域。对于这些增强子,根据区域内对应的Med1这种转录激活通用辅助因子的chip_seq reads的密度进行排序,发现呈现两极分化趋势,示意如下
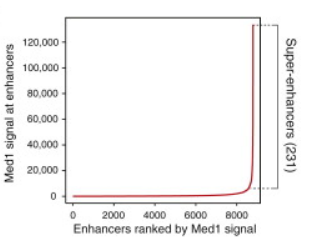
其中绝大部分的增强子对应的Med1的水平都很低,少部分增强子对应的Med1的水平非常高。除了Med1之外,还比较了其他几种转录因子或者组蛋白修饰的数据
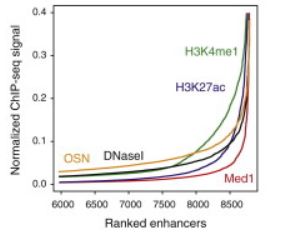
发现Med1的区分效果最佳,根据Med1水平的高低,可以将增强子分为以下两类
-
typical enhancers
-
super enhancers
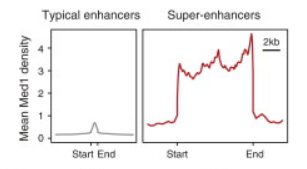
简称TE和SE, 进一步分析发现TE和SE在长度上具有非常明显的区别,SE的长度是TE长度的10倍以上,一个普通的增强子只有几百bp的长度,而超级增强子的长度在几千bp左右。

除了Med1之外,还比较了Qct4等多种转录因子在TE和SE中的分布,结果如下图所示
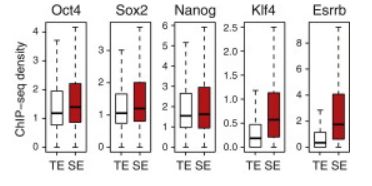
发现在SE中Klf4和Esrrb的分布比TE中更加丰富。对SE区域富集的motif进行分析,结果如下所示

发现富集到了Oct4, Sox2, Klf4等motif。从上述发现和定义超级增强子的过程可以看到,超级增强子的预测过程有以下两个关键点
-
建立在增强子的基础上,可以看做增强子富集的区域
-
相比增强子,超级增强子区域具有更高的转录因子的密度
ROSE这款程序也是根据这两个关键点来识别超级增强子,基本过程示意如下
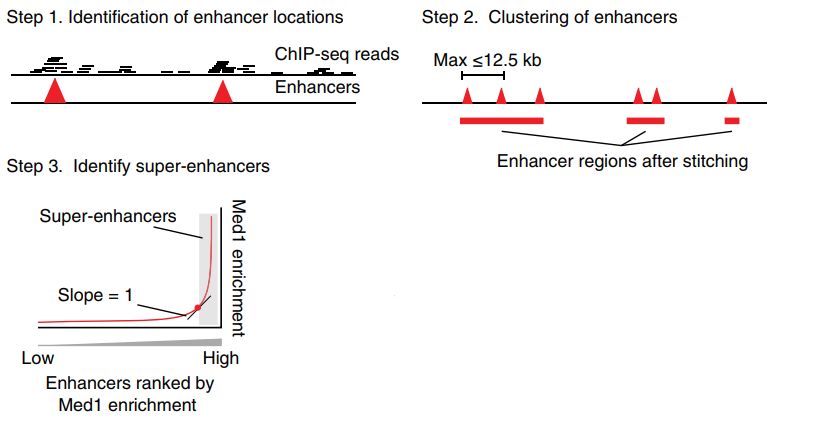
首先识别增强子区域,然后对增强子进行合并,定义一个阈值,将距离小于该阈值的增强子进行合并,最后比较合并后的增强子区域内的reads分布情况来识别超级增强子。
在实际操作过程中,在第一步和第三步可以使用不同的mark, 如下所示

软件基于python编程语言开发,直接从官网下载源代码,解压缩就可以了。源代码中内置了几个物种的注释数据库,存放在annotation文件夹下
annotation/
├── hg18_refseq.ucsc
├── hg19_refseq.ucsc
├── hg38_refseq.ucsc
├── mm10_refseq.ucsc
├── mm8_refseq.ucsc
└── mm9_refseq.ucsc
其实就是从UCSC下载的对应的refGene.txt文件,该软件的基本用法如下
python ROSE_main.py \ -g HG18 \ -i HG18_MM1S_MED1.gff \ -r MM1S_MED1.hg18.bwt.sorted.bam \ -c MM1S_WCE.hg18.bwt.sorted.bam \ -o out_dir \ -s 12500 \ -t 2500
需要注意一定要到软件的安装目录去运行,因为会在运行目录查找annotaton这个文件夹下的物种注释文件。
-g指定参考基因组版本,用于检索对应的物种注释文件;-i指定增强子区域对应的基因组位置,内容如下
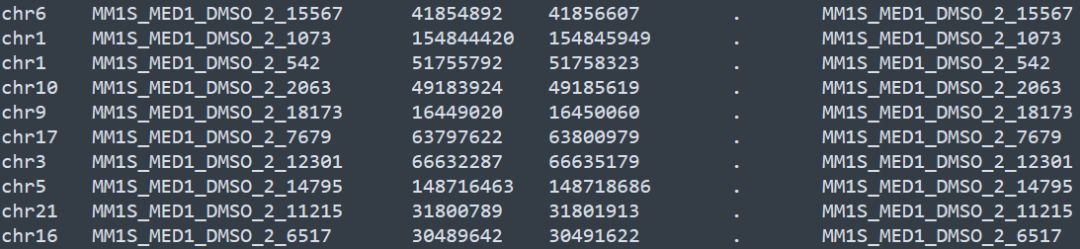
\t分隔的6列,第一列,第三列和第四列指定增强子区域对应的染色体位置,第五列指定正负链信息,.代表不确定,第二列和第六列是一个自定义的唯一的ID, 用来表示增强子的编号。
确定了增强子区间信息之后,接下来就是比较增强子区域内某种mark因子的chip_seq reads的分布情况,-r参数指定chip_seq中IP样本的bam文件,-c指定Input样本的bam文件。
-s指定合并增强子的距离,默认为12.5kb, 小于该距离的两个增强子会合并为一个区间,-t指定距离TSS的距离,如果一个peak与某个转录起始位点的距离小于指定的距离,则有可能是一个启动子,这种潜在的启动子会被过滤掉。
在输出结果的目录会生成很多文件,png文件内容示意如下
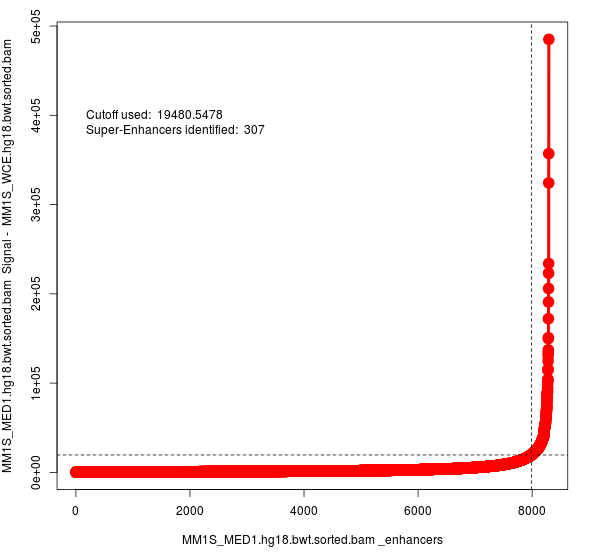
AllEnhancers.table.txt和SuperEnhancers.table.txt分别表示所有增强子和超级增强子的信息,文件内容类似,示意如下
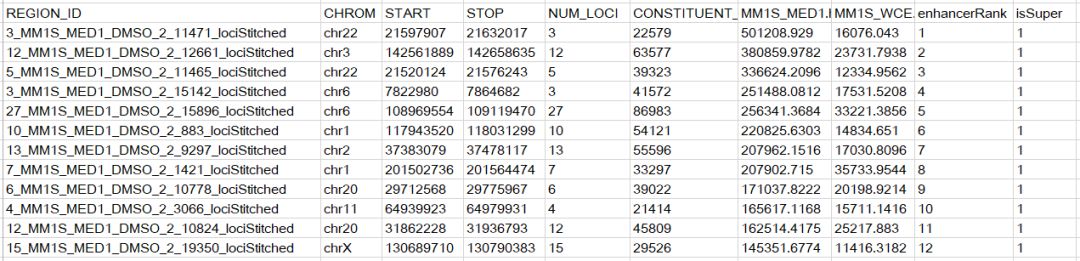
dbSUPER和SEdb这两个超级增强子数据库都是使用h4K27ac组蛋白修饰作为mark来识别超级增强子,可以借鉴这个思路来识别超级增强子。
- 本文固定链接: https://maimengkong.com/zu/1386.html
- 转载请注明: : 萌小白 2023年3月2日 于 卖萌控的博客 发表
- 百度已收录
