全基因组测序(Whole Genome Sequencing),简称WGS,目前默认指的是人类的全基因组测序。所谓全(Whole),指的就是把物种细胞里面完整的基因组序列从第一个DNA开始一直到最后一个DNA,完完整整地检测出来并排列好。因此这个技术几乎能够鉴定出基因组上任何类型的突变。这种从头测序和组装通常应用于没有参考基因组可用或可用参考质量较差的生物体。

之前未测序的基因组必须在测序后通过从头方法组装。然后,该组件可用于其他分析,并为未来的重新测序项奠定基础。
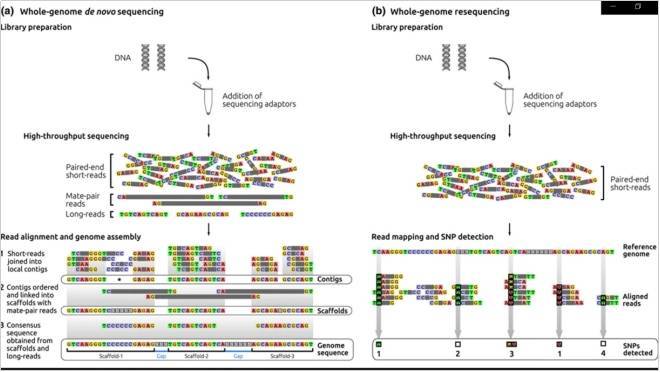
当参考基因组序列可用时,通常会执行重新测序。测序读数与参考基因组对比,以确定基因组中 特定读数最匹配的位置。
重测序通常用于探索个体、家庭和群体的遗传变异,特别是在人类遗传疾病方面。这些研究中对测序深度的要求取决于感兴趣的变异类型、疾病模型和感兴趣区域的大小。
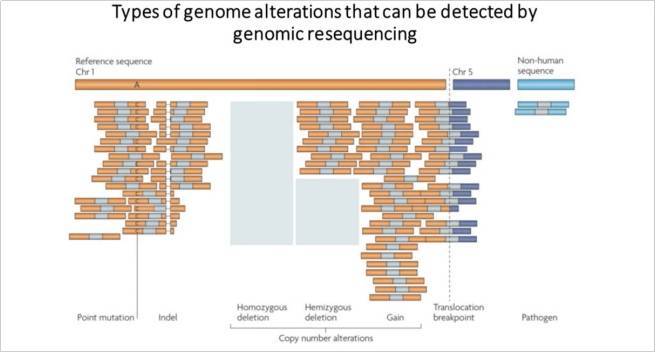
重测序可以揭示单核苷酸多态性、小插入或缺失、结构变异和拷贝数变异。当然,特定研究的设计取决于所讨论的生物学假设,与孟德尔病或癌症体细胞突变研究相比,人口研究使用不同的测序策略。
此外,靶向重测序方法允许在测序广度和样本数量之间进行权衡:在更小的基因组区域上,以相同的成本,更多的样本,可以被测序到相同的深度。
在这里,我们在这些不同的变异类型和疾病模型的背景下讨论全基因组测序 (WGS) 相对于靶向重测序方法(包括 WES)的优点。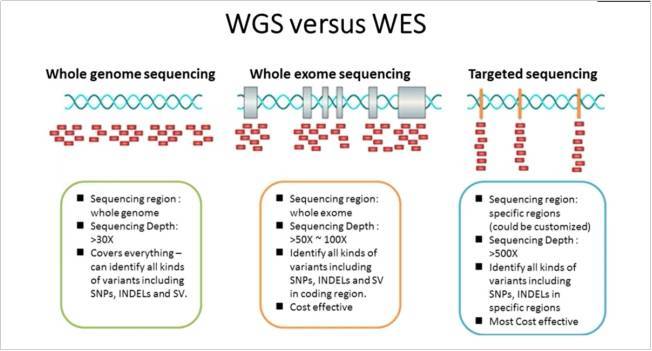
高深度 WGS 是 DNA 重测序的“黄金标准”,因为它可以在编码蛋白质的人类基因组的少数 (1.2%)和其余大多数非编码蛋白质中查找所有的变异类型,包括SNV、插入缺失、结构变异和CNV-编码序列。
WES 专注于检测蛋白质编码基因中的 SNV 和插入缺失以及如microRNA 序列等其他功能元件 ;因此,它省略了启动子和增强子等调控区域。尽管成本因序列捕获解决方案而异,但 WES 可以比 WGS 便宜一个数量级,以实现大致相等的蛋白质编码外显子覆盖范围。这些降低的成本提供了极大增加样本数量的潜力,这是许多研究的关键因素。然而,WES 有各种限制,如下所述。
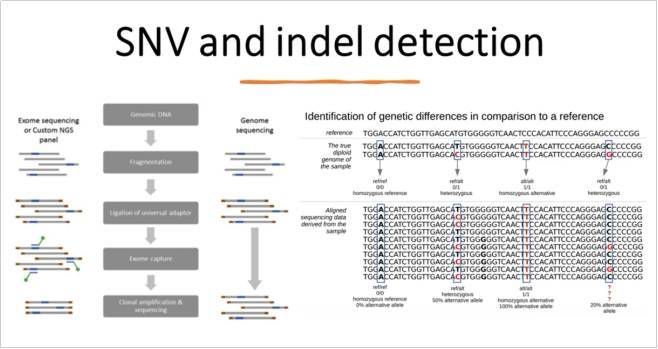
早期的基因组重测序研究特别关注两类最常见的序列变异,即 SNV 和小插入缺失。 第一个使用 Illumina 短读长技术测序的人类基因组表明,尽管几乎所有纯合 SNV都以 15 倍的平均深度处检测到,但要检测相同比例的杂合 SNV,则需要平均 33 倍的深度。因此,超过 30 倍的平均深度迅速成为事实上的标准。
尽管读取质量主要由测序技术决定,但覆盖深度的均匀性也会受到样品制备的影响。在通过 PCR 进行 DNA 扩增期间引入的 GC 偏差已被确定为覆盖率变化的主要来源。消除 PCR 扩增可提高基因组高 GC 区域的覆盖率并减少重复读数。
在 WES 中,序列捕获探针杂交效率的差异可能再次归因于 GC 含量变化,这可能会导致目标区域几乎没有或没有覆盖。覆盖的均匀性也将受到重复或低复杂度序列的影响,这些序列要么限制诱饵设计,要么导致脱靶捕获。
此外,与 WGS 不同,WES 仍常规使用 PCR 扩增,必须对其进行仔细优化以减少 GC 偏差。由于覆盖范围的变化增加,需要更大的平均读取深度才能实现与 WGS 相同的覆盖范围,并且需要 80 倍的平均深度才能覆盖 89.6%-96.8% 的目标碱基。
所有 WES 试剂盒都容易出现参考偏差,这是由与参考序列匹配的捕获探针引起的,因此倾向于优先富集杂合位点的参考等位基因; 这种偏差会产生假阴性SNV 调用。
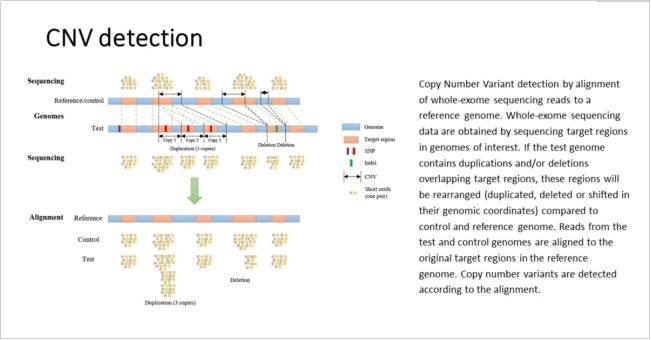
我们可以使用分析覆盖深度的方法从 WGS 和 WES 数据中检测 CNV。这些方法根据基因组坐标堆积对齐的读数,然后计算窗口中的读取,以提供整个区域的平均深度。然后可以从基因组区域的平均深度的变化中推断出拷贝数的变化。
在 WGS 中,平均深度低至 0.1 倍即可获得合理的特异性。然而,灵敏度、断点检测和绝对拷贝数估计都随着读取深度的增加而提高。
无论平均读取深度如何,即使在对 GC 偏差和“可映射性”进行校正之后,覆盖深度方法也容易受到由于覆盖范围的局部变化而被调用的误报,并且需要跨样本调用来减少这种影响。
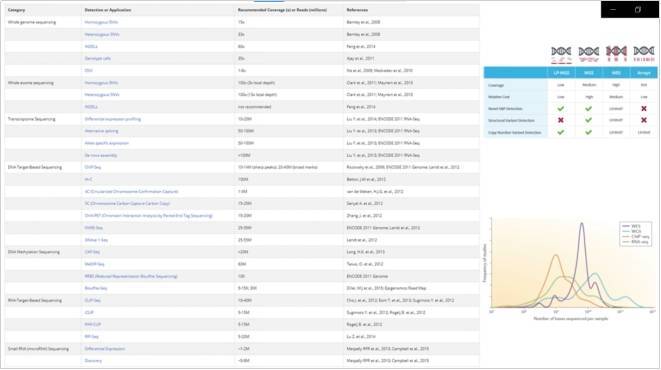
与准确调用单个基因组中的 SNV 和插入缺失所需的深度相比,群体基因组学研究受益于样本数量和测序深度之间的权衡,其中许多基因组在低深度(例如,400 个样本在,4倍) 及其变体在所有样本中被联合调用。
对单个低深度基因组的变异调用具有很高的假阳性率,但通过跨样本组合信息可以减轻这种情况。这种方法以深度测序的测序成本的一部分提供了检测常见变异的良好功能。
实际上,即使是超低覆盖率测序(即以 0.1-0.5倍 测序)也能捕获与单核苷酸多态性 (SNP) 阵列几乎一样多的常见变异(即等位基因频率 >1% 的变异)。
相反,可靠地鉴定高度非整倍体基因组或异质细胞群(例如来自肿瘤的细胞群)中的变异需要比来自正常组织更大的覆盖深度。 有限感兴趣区域的靶向富集和超深度测序(即 1,000 倍测序)可用于研究癌症样本中的克隆进化,其中特定变体存在于 <1% 的细胞群中。
鉴定疾病的新发或隐性变异通常最好通过对亲子三重奏进行测序来实现。在这种情况下,建议为每个家庭成员获得相同的测序深度,以最大限度地减少先证者的假阳性和父母的假阴性。- 本文固定链接: https://maimengkong.com/zu/1230.html
- 转载请注明: : 萌小白 2022年10月4日 于 卖萌控的博客 发表
- 百度已收录
