最早接触PCR是在做科创和本科毕业论文相关实验的时候,通过扩增野生型菌株和突变体菌株的某个基因,然后通过AS-PCR方法,设置温度梯度,筛选一对能够区别S和M菌株的特异性引物。
那个时候对PCR有一个大概的影响,其实在操作方面是没什么问题的。但是仔细想起来,和其他实验一样,都是一知半解,对PCR亦是如此。因此,我觉得有必要重新学习一下PCR相关的各种技术和原理。
第一部分——PCR原理及各种衍生技术
【进入正题】
PCR(polymerase chain reaction)聚合酶链式反应,又称体外DNA扩增技术,在1985年由美国Cetus公司的Kary Mullis首创,可以将微量目的DNA片段扩增一百万倍以上。Kary Mullis本人因此获1993年诺贝尔化学奖。

前面分享过PCR之歌,这里在系统整理PCR相关知识之前,重温一下。
一、PCR的原理
在DNA聚合酶催化下,以母链DNA为模板,以特定引物为延伸起点,通过变性、退火、延伸等步骤,体外复制出与母链模板DNA互补的子链DNA的过程。
聚合酶链式反应用于扩增一小段已知的DNA片段,可能是单个基因,或者仅仅是某个基因的一部分。与活体生物不同的是,PCR只能复制很短的DNA片段,通常不超过10kbp。DNA是双链分子,因此用互补DNA双链的构造单位(核苷酸)来度量其大小,单位为碱基对(base pair, bp)。
二、PCR反应成分
|
template |
过多:非特异性条带增加。 过少:PCR产量降低。 |
|
primer |
预扩增核酸片段两端的已知序列,决定特异性。 偏高:非特异产物扩增及错配,增加引物之间形成引物二聚体,产量降低。 偏低:产量降低。 |
|
polymerase |
耐高热 偏高:引物非特异产物的扩增。 偏低:产物量降低。 |
|
dNTP |
dATP、dGTP、dCTP、dTTP 过高:加快反应速度,还可增加碱基的错误掺入率和室验成本。 过低:反应速度下降,可提高实验的精确性。 |
|
buffer |
Mg2+ Taq酶具有Mg2+依赖性,显著影响反应的特异性和扩增片段的产量。 过量:能增加非特异扩增并影响产率。 过低:则酶活性显著下降。 10~50 mM Tris- Cl (pH8.4) 维持Taq酶作用环境的偏碱性 25~50 mM KCl 促进引物退火,>50 mM会抑制Taq酶的活性。 100μg/ml牛血清白蛋白(BSA) 对酶有一定的保护性,如质量不好将起相反的作用,建议使用乙酰化的BSA。明胶、Tween-20、二硫苏糖醇(DTT)也有类似作用。 |
上述也就是现在广泛使用的mix的主要成分。
三、PCR反应基本步骤
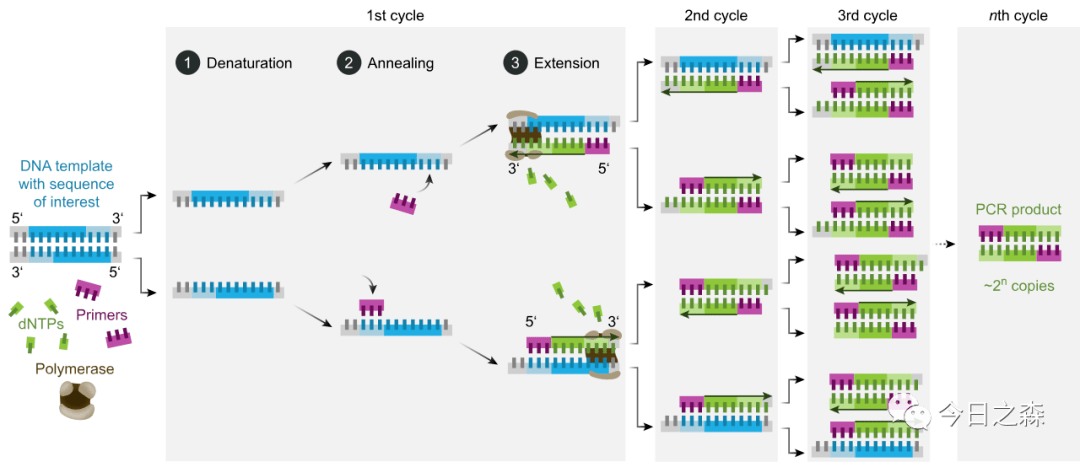
一般的聚合酶链式反应由20到35个循环组成,每个循环包括以下3个步骤:
- 变性(Denaturation): 利用高温(93-98℃)使双链DNA分离。高温将连接两条DNA链的氢键打断。在第一个循环之前,通常加热长一些时间以确保模板和引物完全分离,仅以单链形式存在。该步骤时间1-2分钟,接下来PCR仪就控制温度进入循环阶段。
- 退火或称接合,复性(Annealling): 在DNA双链分离后,降低温度使得引物可以结合于单链DNA上。此阶段的温度通常低于引物熔点5℃。错误的退火温度可能导致引物不与模板结合或者错误地结合。该步骤时间1-2分钟。
- 延伸(Extension): DNA聚合酶由降温时结合上的引物开始沿着DNA链合成互补链。此阶段的温度依赖于DNA聚合酶。该步骤时间依赖于聚合酶以及需要合成的DNA片段长度。传统的Taq估计合成1000bp/min、较新的Tbr(来自于嗜热菌Thermus brockianus)约40秒、商业公司生产的融合型聚合酶仅需约10-15秒。
四、PCR反应条件优化
1、变性温度和时间:
保证模板DNA解链完全是保证整个PCR扩增成功的关键。
加热90~95°C, 30~60s,再复杂的DNA 分子也可变性为单链。
温度过高或高温持续时间过长,可对Taq酶活性和dNTP分子造成损害。
2、复性温度和时间:
PCR扩增特异性取决于复性过程中引物与模板的结合。
复性温度越高,产物特异性越高。复性温度越低,产物特异性越低。
需根据引物的Tm值具体设定。
3、延伸温度和时间:
一般位于Taq酶最适作用温度70~75°C之间。引物小于16个核苷酸时,过高的延伸温度不利于引物与模板的结合,可以缓慢升温到70~75°C。
延伸反应时间,可根据待扩增片段的长度而定,小于1kb, 1min足够;大于1kb需加长延伸时间。Taq酶可根据1kb/min增加时间。
这里需要注意,延伸时间过长可能出现非特异扩增。因此需要设置恰到好处的延伸时间。
4、循环数:
其他参数选定后,PCR循环次数主要取决于模板DNA的浓度。
理论上说20〜25次循环后,PCR产物的积累即可达到最大值,实际操作中由于每步反应的产率不可能达到100%,因此不管模板浓度是多少,20~30次是比较合理的循环次数。循环次数越多,非特异扩增增加。
五、PCR延伸技术
由PCR延伸而来的技术很多,这里只介绍日常实验中常用到的几种。
1.touchdown PCR
降落PCR,主要用于PCR的条件的优化。在许多情况下引物的设计使得PCR难以进行,例如特异性不够易错配等。退火温度过高会使PCR效率过低,但退火温度过低则会使非特异扩增过多。
因此,前面几个循环的起始退火温度设定为比引物的最高熔解温度(Tm)再高几度。前几循环温度逐渐下降至设定的最终Tm。通过较高温度获得特异性匹配较高的模板后,再以较低温度高效率扩增。
2.RT-PCR
逆转录-聚合酶链反应(Reverse Tranion-Polymerase Chain Reaction,RT-PCR)的原理是:提取组织或细胞中的总RNA,以其中的mRNA作为模板,采用Oligo(dT)或随机引物利用逆转录酶反转录成cDNA。再以cDNA为模板进行PCR扩增,而获得目的基因或检测基因表达。
3.real-time PCR/quantitative PCR
实时荧光定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。
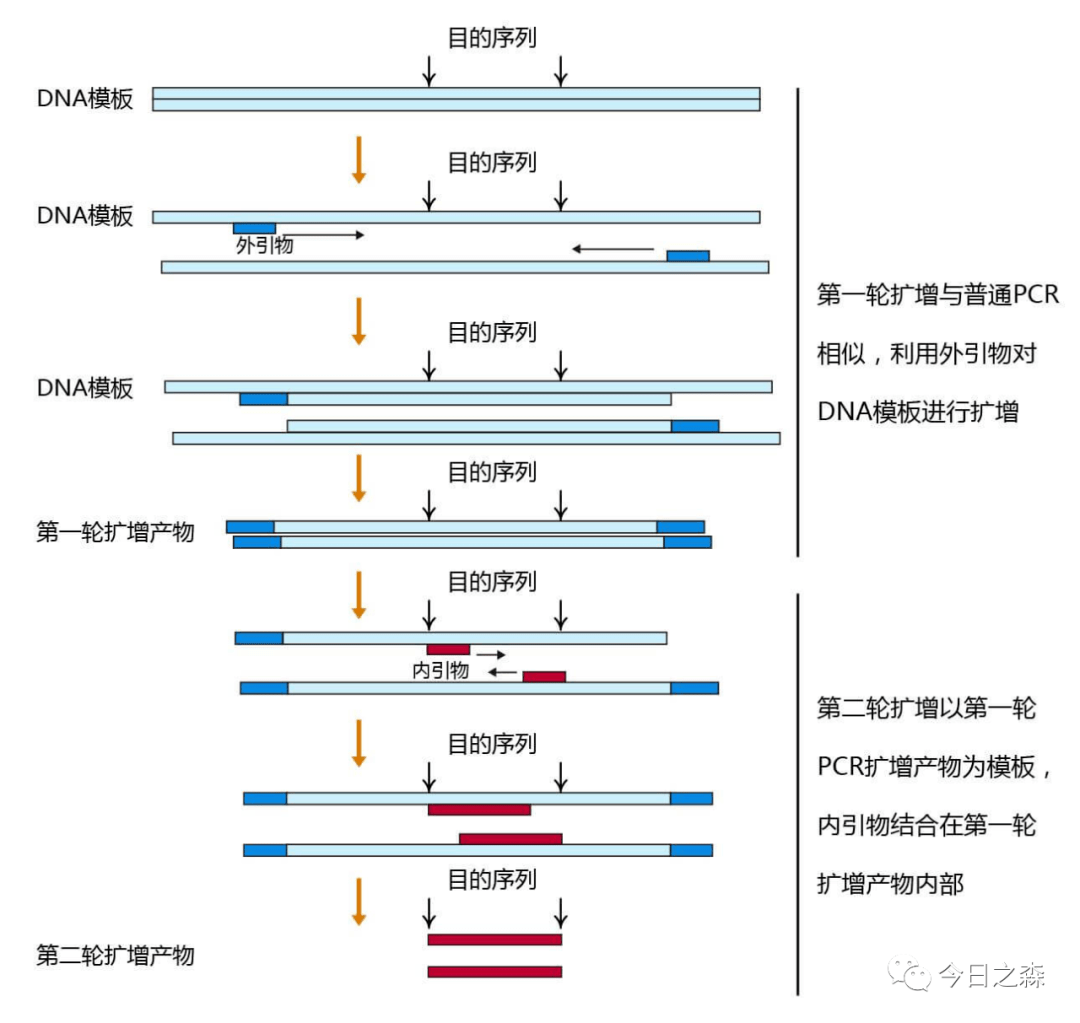
4.nested PCR
先用低特异性引物扩增几个循环以增加模板数量,再用高特异性引物扩增。
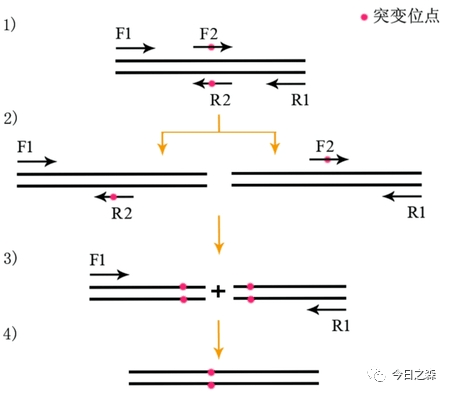
5.SOE PCR
重叠延伸PCR分为2种:用重叠延伸PCR做定点突变 和 用重叠延伸PCR做序列缺失突变,即重叠延伸PCR技术(gene splicing by overlap extension PCR,简称SOE PCR)
5.1 用重叠延伸PCR做定点突变(由于原理简单,直接上图)
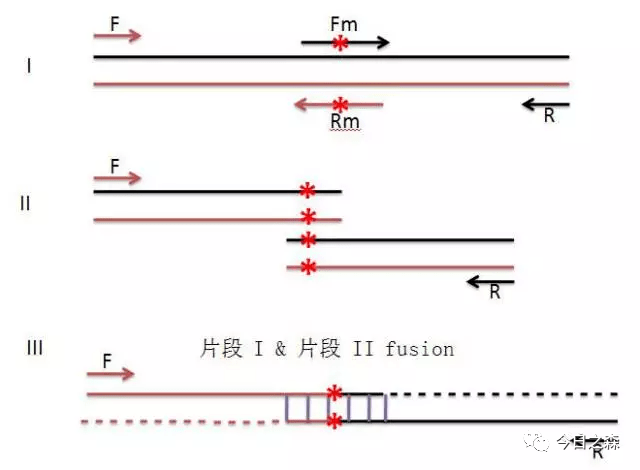
该步一定要用pfu酶,不能用Taq酶,因为Taq酶容易在PCR产物末端加A,会造成产物移码突变。
5.2 用重叠延伸PCR做序列缺失突变

6.高GC含量PCR
具有高GC含量(>65%)的DNA模板由于G和C碱基间的强氢键影响,比较难以扩增。富含GC的序列同时也涉及二级结构。因此,富含GC的序列可导致DNA聚合酶沿模板扩增时“卡顿”并干扰DNA合成。
为了扩增高GC含量的片段,双链模板必须解离,以便引物与模板结合,并使DNA聚合酶能够读取到序列。为了克服强GC相互作用,最常用的方法是使用DMSO等PCR添加剂或辅助溶剂来帮助DNA变性。然而,这些试剂通常会降低引物的 Tm,所以退火温度也需进行相应的调整。
高合成能力的DNA聚合酶由于与模板的结合能力更强,有利于完成高GC含量PCR。超高热稳定性DNA聚合酶也有利于高GC含量PCR,因为较高的变性温度(如,使用98°C代替95°C)可能会促进双链解离和PCR扩增。
7.AS-PCR
等位基因特异性PCR( allele specific PCR,AS-PCR ),是指利用引物与模板之间的碱基错配可以有效地抑制PCR反应,进而达到模板区分(等位基因区分)的目的。
由于PCR过程中引物延伸是3'端开始的,所以3'末端的碱基对引物的延伸来说处于至关重要的位置。如果这个碱基与模板互补,则引物能不间断延伸,PCR可以正常进行,得到特定长度扩增带,反之,则不能延伸。所以只要将与正常等位基因所不同的那个突变碱基安排在引物3'最末端,当用某一含突变序列的引物进行PCR时,如果得到特异条带,表明被测基因含有该种突变。没有特异扩增带出现,则表示没有这种突变。
注意:这里由于仅仅利用了引物3'末尾碱基的错配,因此需要摸索一个合适的Tm,才能达到检测目的。
【Reference】
Mullis, Kary B. et al. "Process for amplifying, detecting, and/or-cloning nucleic acid sequences" 美国专利第4,683,195号
第二部分——引物设计
【进入正题】
前面提到的几个探究蛋白互作相关的实验,其实第一步都需要构建合适的质粒,那么就少不了设计靠谱的引物了。
通常来说,引物的设计需要遵守一些基本的原则。这些原则很细,很多,也很繁琐。但实际上对于常规的引物,并不需要考虑太多,达到实验目的即可,更多的情况下需要灵活选择。
一般来说,引物设计需要遵循以下原则:
1. 引物的长度一般为15-30 bp,常用的是18-27 bp,但不应大于38,因为过长会导致其延伸温度大于74℃,不适于Taq DNA聚合酶进行反应。
2. 引物序列在模板内应当没有相似性较高,尤其是3’端相似性较高的序列,否则容易导致错配。引物3’端出现3个以上的连续碱基,如GGG或CCC,也会使错误引发机率增加。
3. 引物3’端的末位碱基对Taq酶的DNA合成效率有较大的影响。不同的末位碱基在错配位置导致不同的扩增效率,末位碱基为A的错配效率明显高于其他3个碱基,因此应当避免在引物的3’端使用碱基A。另外,引物二聚体或发夹结构也可能导致PCR反应失败。5’端序列对PCR影响不太大,因此常用来引进修饰位点或标记物。
4. 引物序列的GC含量一般为40-60%,过高或过低都不利于引发反应。上下游引物的GC含量不能相差太大。
5. 引物所对应模板位置序列的Tm值在72℃左右可使复性条件最佳。Tm值的计算有多种方法,如按公式Tm=4(G+C)+2(A+T)。
6. ΔG值是指DNA双链形成所需的自由能,该值反映了双链结构内部碱基对的相对稳定性。应当选用3’端ΔG值较低(绝对值不超过9),而5’端和中间ΔG值相对较高的引物。引物的3’端的ΔG值过高,容易在错配位点形成双链结构并引发DNA聚合反应。
7. 引物二聚体及发夹结构的能值过高(超过4.5kcal/mol)易导致产生引物二聚体带,并且降低引物有效浓度而使PCR反应不能正常进行。
8. 对引物的修饰一般是在5’端增加酶切位点,应根据下一步实验中要插入PCR产物的载体的相应序列而确定。
但是在实际设计过程中,往往很难同时满足以上条件,因此只需要在设计引物时尽可能满足即可,没必要太过纠结。
同时需要注意,各种模板的引物设计难度不一,具体情况需要具体看待。有的模板本身GC含量偏高或偏低,导致找不到各种指标都十分合适的引物;在用作克隆目的的PCR因为产物序列相对固定,引物设计的选择自由度较低。在这种情况只能退而求其次,尽量去满足条件。
下面主要介绍以下两方面的引物设计。
1 点突变引物设计
2 qPCR引物设计
(常规引物的设计思路在《实验技术(1)——载体构建相关》已经提到,这里不再重复。)
1 点突变引物设计
在蛋白质功能研究过程中常常对其关键的酶活位点、修饰位点进行突变,通过点突变来研究基因中发挥关键功能的是哪一个结构域,甚至具体到哪一个氨基酸。因此就需要设计点突变引物。
点突变主要的原则就是通过尽量突变较少的碱基数目而使氨基酸发生更改。由于氨基酸密码子第三位具有简并性,一般不需要进行突变,因此至多突变两个碱基即可。
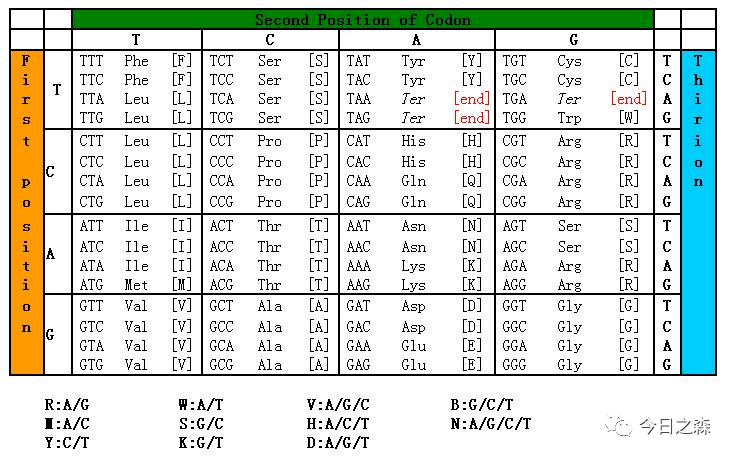
1.1单点突变引物设计
比如,将下图中的Ser突变为Ala。
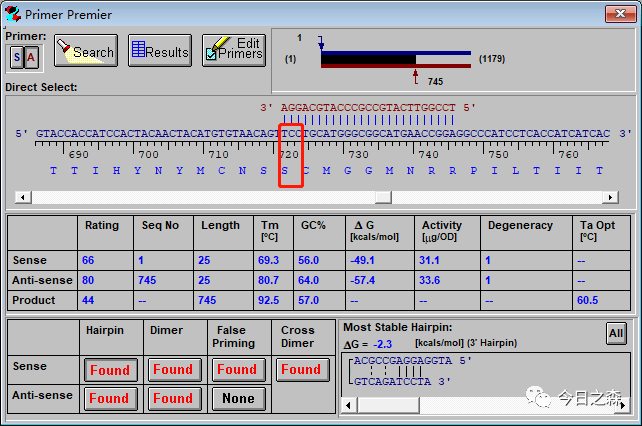
首先,查询编码Ala的密码子:为GCA/GCT/GCG/GCC,根据改动最小原则,我们选择将其突变为GCC,则只需要改动1个碱基,即TCC→GCC。
设计引物(分两种情况):
情况一:构建点突变质粒
只需下面这一对引物即可。
F-S241A:TGTACACATTGTCACGGACGTACCCGCCGT
R-S241A:ACGGCGGGTACGTCCGTGACAATGTGTACA
可以看到黄色部分为需要突变的氨基酸,红色的碱基为突变碱基,两边各延长一部分完全与模板互补配对的碱基。
但是发现两条引物完全互补配对,看似违反了前面所讲的原则,但实际上是针对质粒点突变精妙的设计。两条引物有一定几率与模板匹配,此时便会按5‘-3’方向完整扩增一圈整个环状质粒,最终形成突变质粒的构建。
情况二:只对目的基因进行点突变
则除了需要情况一中的两条引物,还需要在目的基因两端选取F和R。如下图:
1.2同时对多个位点进行点突变
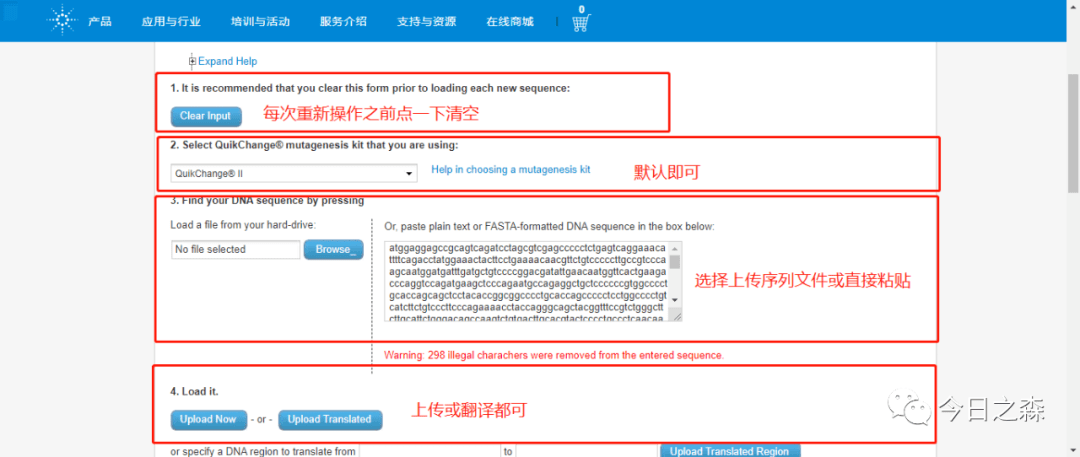
针对单点突变,手动选取即可完成,但对于多位点突变就不能蛮干了。这里分享一个在线点突变引物设计的网址:
该网站可以设计单点和多点突变引物以及删除或者插入一段DNA序列(插入时只能插入小的DNA片段)。可以同时设计针对7个氨基酸的突变引物。
进入页面:
参数说明:
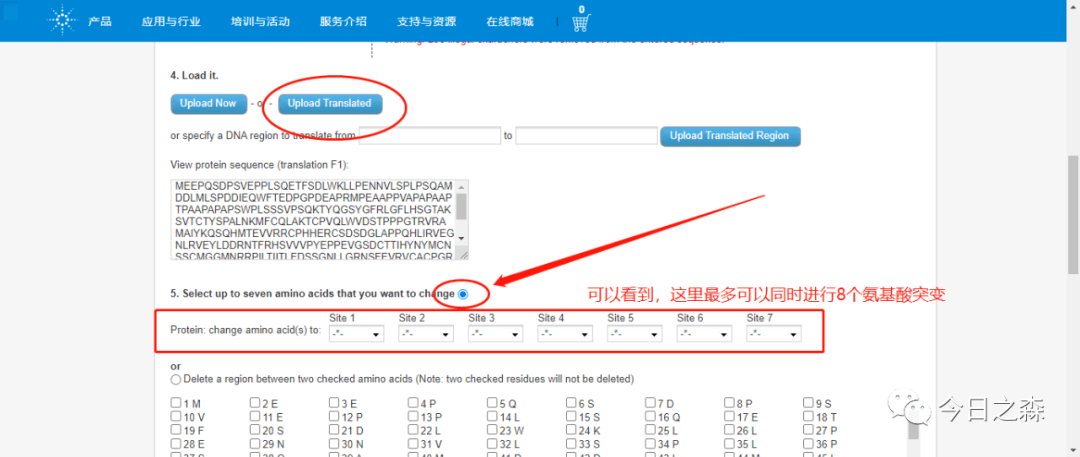
比如,我们对以下3个位点进行点突变。
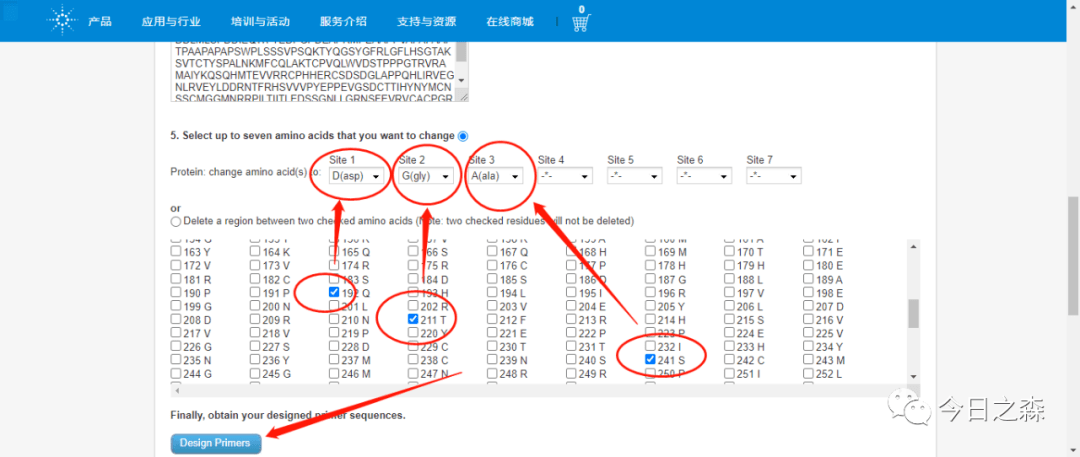
从结果可以看到引物的长度、Tm值及引物与模板相互匹配的序列。

1.3缺失一段序列
比如缺失下图中67-114之间的序列, (注意:第67位和114位这两个氨基酸不会被删除,实际删除66-113这段氨基酸序列)。
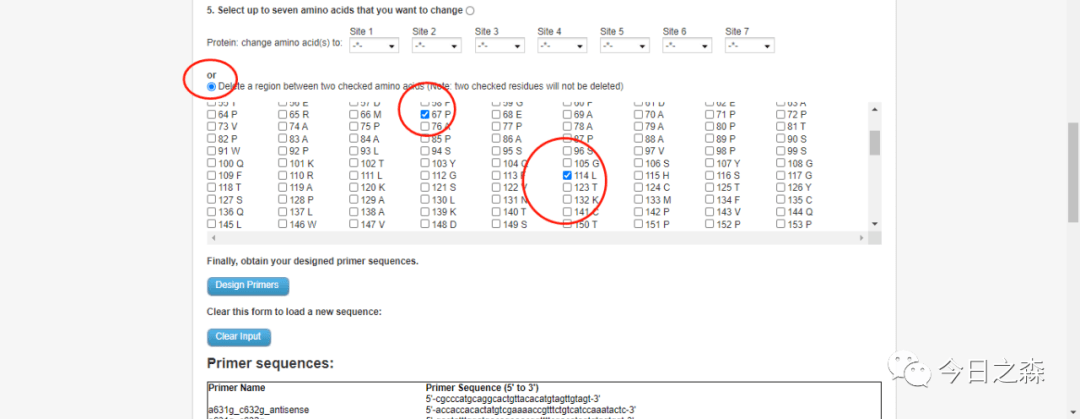
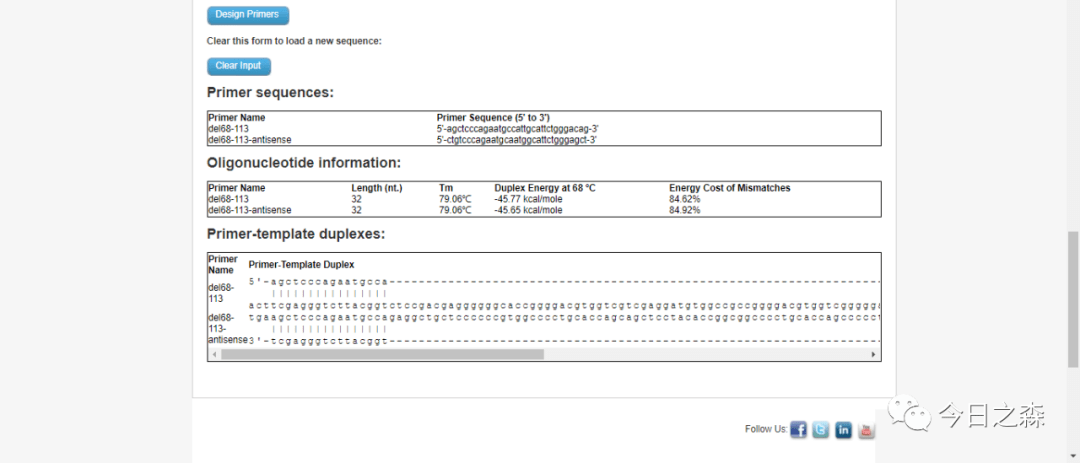
小结:一般在单点突变和缺失一段序列时,成功率比较高;多点突变时可能就需要多次尝试并检测了。
2 qPCR引物设计
Q:qPCR引物设计的问题点:
A:SYBR Green与双链DNA结合后会产生荧光,如果反应体系中有非特异性扩增或者引物二聚体存在,也将被检测到,导致实验结果的不准确。
因此设计合适的qPCR引物就很关键。
qPCR引物设计的一般原则:
- 引物应在核酸系列保守区内设计并具有特异性。
- 扩增产物长度在80-150 bp。最长不要超过300 bp。
- 产物不能形成二级结构。
- 引物长度:一般在17-25碱基之间,上下游引物不宜相差太大。
- 引物自身避免形成发卡结构。
- 引物之间避免形成引物二聚体。
- 引物 G+C含量在40%~60%之间,45-55%最好。
- 引物Tm值在58-62度之间,上下游引物Tm值不宜相差太大,最好不要超过5度。
其实这里qPCR引物的设计和常规PCR的设计也是大同小异。和常规引物设计一样,并不能死磕原则,灵活选择更为重要。
这里可以使用以下软件和在线网站进行设计:
1.Primer Premier 5.0
设计方法和常规引物设计一样,只需根据实验需求设置几个必要参数即可。这里不进行演示。
2.NCBI Primer blast:
3.Primer3:
下面对NCBI Primer blast和Primer3进行简单介绍。
NCBI Primer blast
同样,我们选择P53这个基因。点击Pick Primer进入NCBI Primer blast界面。
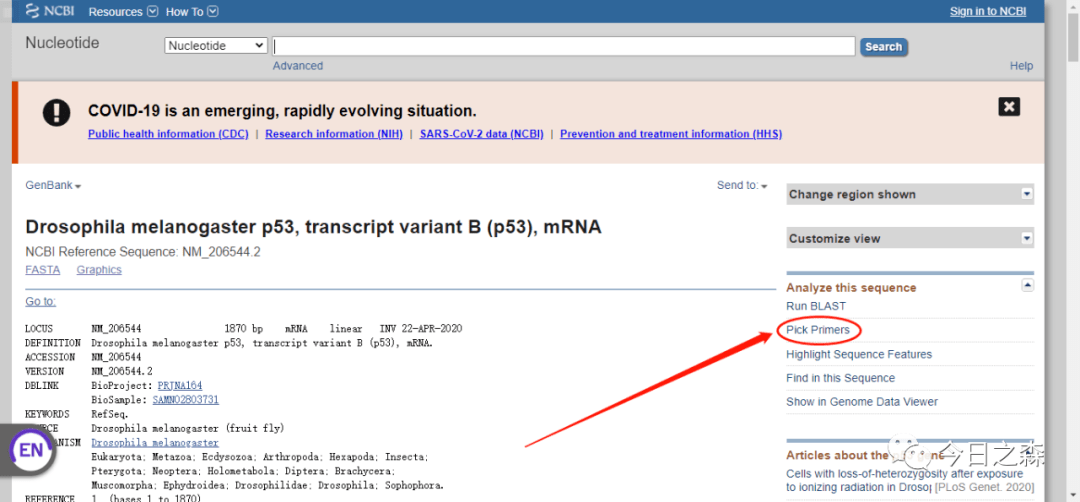
可以看到,上一步选择的p53序列已经加载进来。
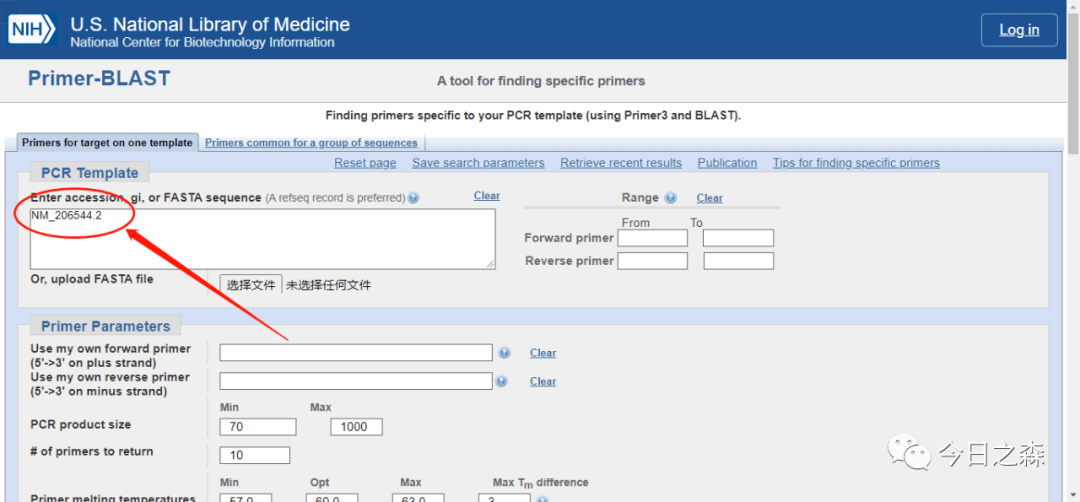
接下来看一下界面中几个重要的参数设置。
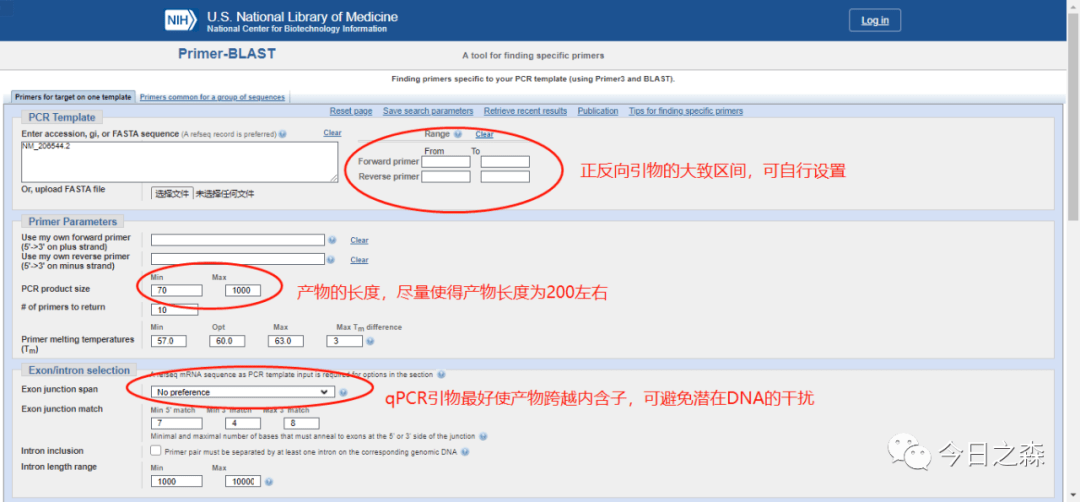
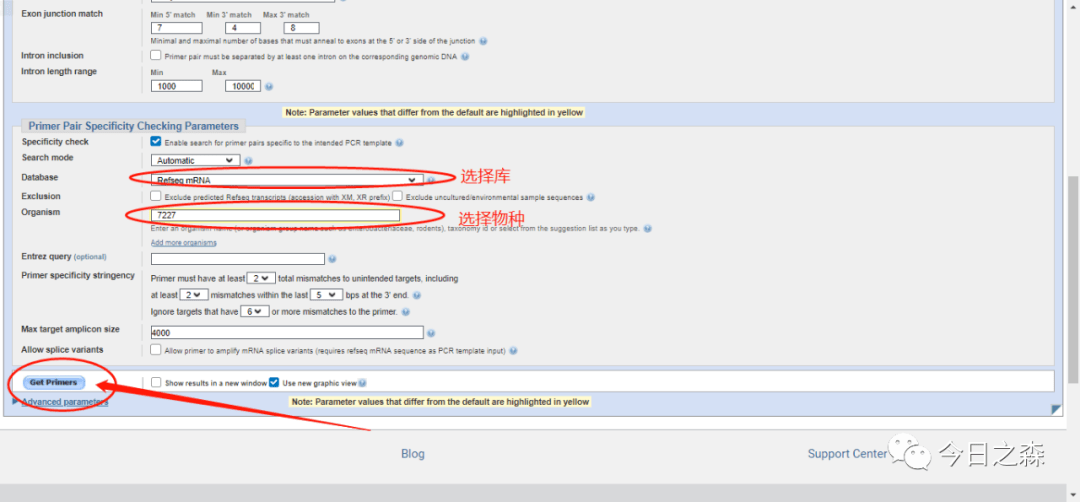
结果如下:
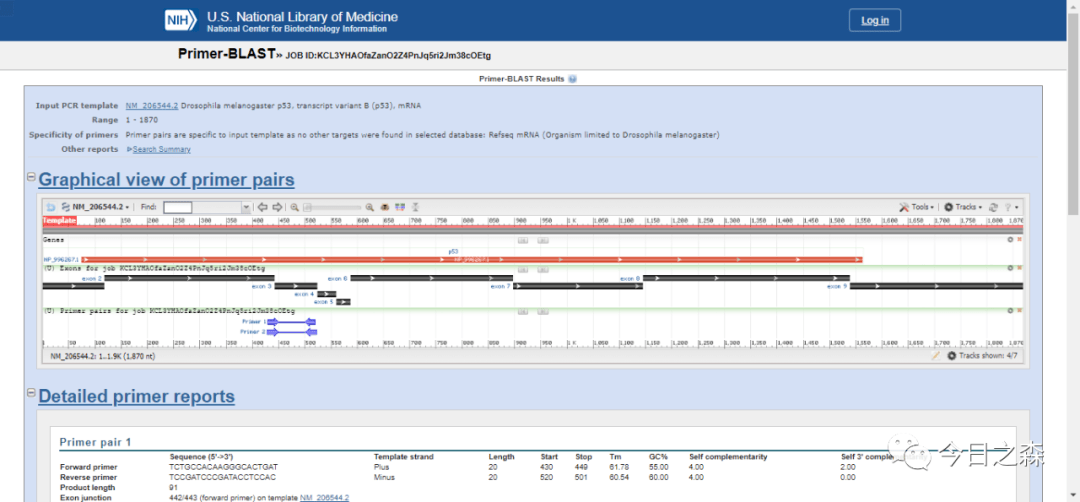
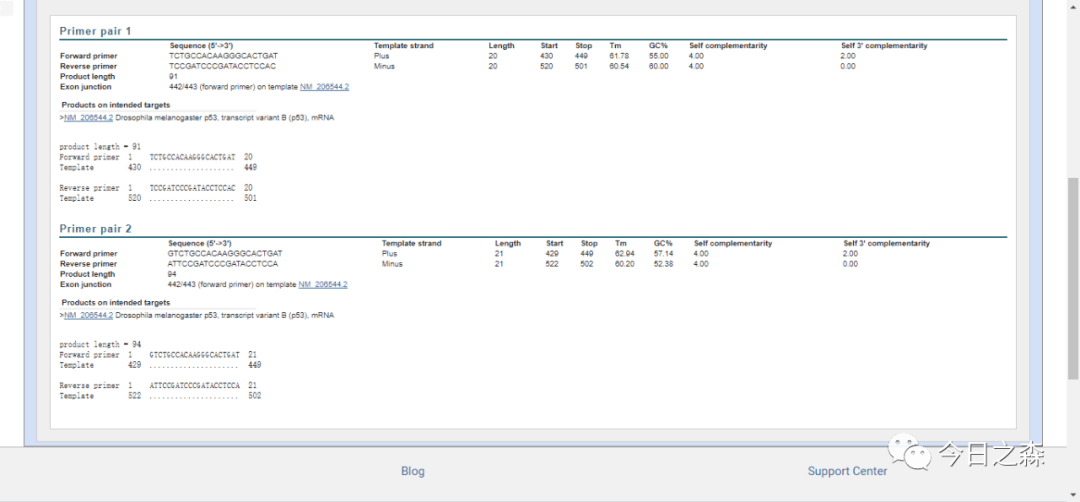
最后,选择最优引物即可。
Primer3
Primer3属于傻瓜式操作,各参数已自动设定,基本无需更改,粘贴序列后直接提交即可。
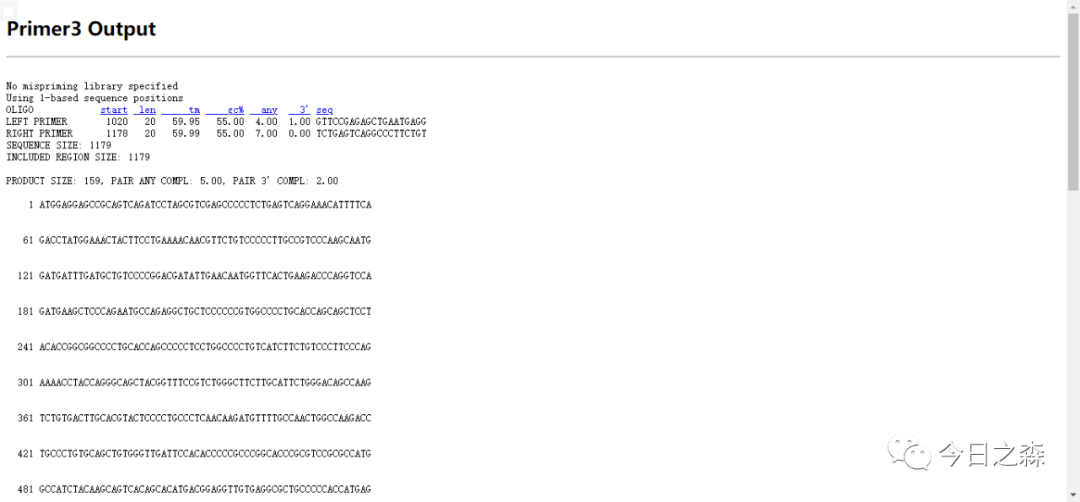

但这里需要思考一个问题:
是否排名第一的引物对一定最优呢?不一定。由于这些引物是未考虑非特异性扩增的。因此在得到候选引物对后,最好还是灵活选择。以便得出最优的引物对,确保实验的准确性。
除了上述在线网址外,给大家推荐以下软件和在线网址,助力解决点突变、qPCR等引物设计的各种问题。具体的使用问题各位可以自行摸索,都很简单,基本上打开就可以使用
1.BioXM
2.http://www.bioinformatics.org/primerx/index.htm
3.https://biodb.swu.edu.cn/qprimerdb/
(qPCR引物数据库qPrimerDB,收录了包括66种重要植物(拟南芥、水稻、玉米、大豆、马铃薯等)等在内的147个物种共3331426个基因的51091785对qPCR引物。)
转自:科研人直通车- 本文固定链接: https://maimengkong.com/kyjc/1229.html
- 转载请注明: : 萌小白 2022年10月4日 于 卖萌控的博客 发表
- 百度已收录
