BSA在遗传学、基因组学和育种中的应用
摘要
以前生物学分析需要基于样本群体中的所有个体开展,但是混合样本分析(Bulked sample analysis, BSA)这种通过挑选个体并进行混合分析的方法渐渐在遗传绘图方面得到了广泛运用,方式包括构建双亲本遗传群体、测序重要基因突变体或是对极端突变体进行GWAS分析等。与传统的基于整个样本群体的分析相比,BSA显著降低了测序的规模(数据量)和花费。混池可以通过在任何群体中挑选极端性状(或者代表性性状)个体来构建,混池测序可以使用单独的分子标记、芯片和高通量测序在包括DNA、RNA和蛋白质在内的所有水平进行研究。BSA的分析能力受到群体大小、极端性状个体选择策略、测序策略、目标性状的遗传结构以及标记密度等因素的影响。BSA可以通过标记开发、标记辅助选择育种、选择性基因分型以及农作物基因组学等方式提升作物育种的研究水平。我们将从BSA在遗传学、基因组学、作物育种等方面的应用以及未来的发展方向展开讨论。
简介
遗传学、基因组学和作物育种等方面的生物学研究(例如遗传图谱绘制等)以前通常需要使用一系列遗传因子(例如分子标记等)对一个群体中所有样本的信息来开展研究。为了保证结果在统计学上的可靠性,通常需要较大的群体和较高的标记密度。对于很多数量性状来说,它们可能受到多个基因、环境等因素的影响,在遗传上较为复杂,为了解析物种的遗传结构或是利用一些重要性状,数量性状位点(QTL)的定位就显得非常重要。然而,传统的分析方法需要对群体中所有个体进行解析,从而非常耗时且花销很大。
为了在保证统计学可靠性的同时简化分析方法并且降低成本,有学者提出了仅使用部分极端表型个体进行选择性分析的方法。更进一步降低成本的方法是从表型分布的两端挑选个体并混池进行分析,例如从两个不同表型的混池中提取DNA进行标记开发,即混合样本分析(Bulked sample analysis,BSA)。之后,有学者对BSA方法进行了修改,使其能够通过较大的群体和较高密度的标记来进行目标基因的定位。从结果上看,这种方法在保证了统计学上可靠的QTL定位能力的同时,极大低降低了基因分型的成本。假设有一个500个个体的群体,从性状分布的两端各取25个个体构建混池,BSA相对于整个群体的分析来说,仅需要0.4%(2/500)的成本。
随着近年来分子育种技术的不断发展,BSA方法也得到了很多改进。DNA混池可以在不同群体中选择极端性状的个体来构建,包括但不限于双亲本构成的遗传群体。例如可以使用主要基因发生变异的个体和它们对应的野生型个体分别组成混池进行分析,也可以使用Mutmap的策略进行分析。而自然群体中也可以通过筛选极端表型个体构成混池并测序来进行全基因组关联分析(GWAS)。在这篇文章中,BSA这个术语包含了遗传育种中所有选择个体并混池分析的方法。我们将BSA定义成从样本群体中挑选部分个体构成具有代表性的混池的分析方式。
在这篇文章中,我们将从遗传学、基因组学和育种等研究的角度对BSA进行描述和评价。我们还将描述相关的样本选择策略、混池策略、以及从DNA、RNA、蛋白质等层面进行分析的策略。最后,我们还将对BSA方法在遗传学、基因组学和作物育种等方面未来的应用进行讨论。
对于任意两组不同的个体组成的混池,只要目标性状在两组之间存在较显著差异,而非目标性状在两组的个体之间随机分布,就可以应用BSA方法进行分析。样本可以从以下两类群体中进行挑选:(i)双亲本或者多亲本遗传群体中的分离群体;(ii)任何群体中的变异个体组成的群体,包括那些遗传背景特别复杂的自然群体。
分离群体
分离群体混池可以从双亲本、三亲本、四亲本或是多亲本杂交群体中进行挑选,这其中也包括那些设计较为复杂的群体例如双列杂交群体、NCD群体、MAGIC群体和NAM群体等。
具备性状差异的双亲本群体在BSA分析中应用最为广泛。这类群体包括F2、F2:3、BC1、RILs和DHs群体。其中RILs群体和DHs群体是由纯合个体组成,在自交后可以保留性状,具备在不同时间和环境下进行验证的能力。
几种测交群体可以从双亲本群体或是多亲本群体中通过与纯合亲本测交构建得到,在这种情况下,需要对测交使用的个体进行基因分型,并对测交后代进行表型分析。双列杂交、NCD I,II,III和三重测交群体也可用于构建测交群体,测交群体可以用于研究杂种优势等重要的遗传学机制。
变异群体
与人工构建的遗传群体不同,变异群体通常来源于自然群体或是突变体库。使用这类群体中现存的差异表型可以绕过构建大型分离群体的步骤。
自然群体包括一个特定物种的近交系(inbred)、种质库(accession)、生态型(ecotype)等,包含目标性状的一系列变异。这类群体通常包含较为复杂的遗传背景,可以用于多个目标性状的研究。由于个体间在非目标性状上也存在较大差异,所以表型鉴定最好能够在受控制的环境下进行。自然群体在植物研究中得到了广泛运用。
通过定向诱导基因组局部突变(target-induced local lesions)方法可以诱变得到大量突变个体,为了让突变覆盖基因组的所有位点,需要得到数量非常庞大的突变个体,非常耗时且非常昂贵。突变体库可以用来寻找罕见突变、进行miRNA功能分析、检测和记录EMS诱变得到的突变等。
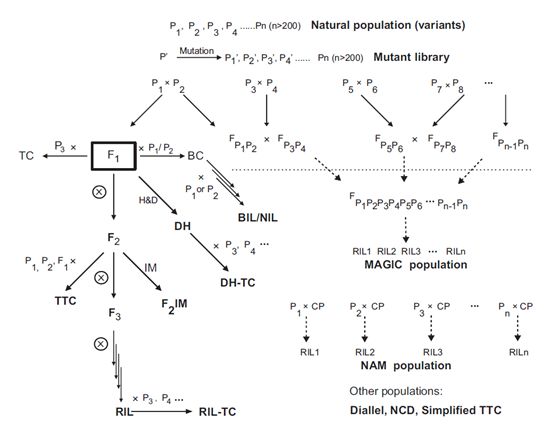
图1 植物群体类型及其相互关系
样本:样本选取、混池构建和混合分析
最开始BSA被用于研究那些受主效基因控制而较少受环境影响的性状,而最近的BSA方法可以更有效地鉴定出对表型有影响的较罕见的次要等位基因。一项模拟研究发现BSA可以用于定位那些对表型影响更小的QTL以及相互连锁或是相互作用的QTL。初始种群大小为3000时,选择两个极端性状各10%的个体,标记密度5cM,我们可以检测到95%表型贡献度1%的QTL。酵母中一项检测微效基因的研究支持了这一结论。
BSA鉴定位点的能力很大程度上依赖于极端性状个体的选择,而这又很大程度依赖于表型鉴定的准确程度,尤其是那些受环境影响较大的性状。为了提升表型鉴定的精确度,降低信噪比是一个非常重要的环节。这可以通过减少土地性质的差异、杂草控制、病害控制、充分的边界隔离、通过实验设计降低重复之间的差异以及通过数据分析方法消除空间倾向等。精确的表型鉴定还依赖于一些新的控制手段,如精确施肥、水分管理、遥感技术等,还有就是准确的表型测量方式。
对于生物或是非生物的胁迫,表型鉴定需要同时在2个差异明显的环境当中进行,或者使用near iso-environments (NIEs),其中一个环境对作物显示出更少的胁迫。胁迫对于作物的影响可以在两个差异环境之间进行比较,也可以使用自然环境作为对照。从中可以计算出一个性状的相对数值,来研究作物对于胁迫的敏感度。适用于NIEs来进行测量的表型包括生物或非生物胁迫(包括抗病或抗干旱)以及农业技术措施(如杂草控制等)。
样本选择
基于性状的样本选择(trait-based sampling)和基于标记的样本选择(marker-based sampling)这两种差异很大的样品选择方式都可以应用于BSA当中。前者选择感兴趣性状中呈现极端表型的个体,而后者基于均匀覆盖分离群体基因组的分子标记,并对目标区域的标记进行基因分型以选择样本。在遗传学中,前者通常用于绘制草图而后者通常用于绘制精细图。不过,BSA通常基于性状进行样本选择。
样本的相关参数会影响BSA的定位能力,尤其是群体大小和混池大小的影响较大。在BSA分析中,群体大小的要求会根据群体类型、标记间距、目标区域重组频率和目标性状的遗传结构不同而发生变化。由于重组频率和基因分型的信息会在不同的群体类型之间有较大差异,要求的群体大小也会存在比较大的差异。
对于那些由微效基因控制的复杂性状,目标基因的其他参数如基因数量,基因效应、基因间互作以及基因在染色体上的相对位置都需要纳入考虑来确定所需的群体大小。为了更高效地确认标记和性状之间的联系,确定群体大小时还需要考虑标记是否可用以及基因分型的成本。基因分型成本下降的话,提升群体大小的空间就更大。
以遗传图谱作为例,挑选多少极端性状个体通常取决于整个群体的大小以及基因效应的大小。对于中小型群体(200-500个体),较适合的极端性状混池大小大约是整个群体的20%-30%。随着群体大小的增长,选择用于QTL定位的样本占比会逐渐减少。对于一个具有较强效果的QTL(表型贡献率在10%-15%以上),每一端的极端性状个体选取需要超过20个(或者在整个群体中选择比大于10%)。对于中效的QTL(表型贡献率在3%-10%之间),每一端的极端性状混池需要在500-1000个体的群体中选取超过50个个体(在整个群体中选择比在5%-10%之间)。对于微效QTL(表型贡献率在0.2%-3%之间),每一端的极端性状混池需要在3000-5000个体的群体中选取100个个体。考虑最佳的选择比例的时候,需要使基因分型和表型鉴定的成本取得平衡。
混池
目标极端性状的选择可能是单端也可能是双端选择,所以对于一个目标性状来说,可能有1个,2个甚至多个混池,提供了不同的比较方式(图2)。有4种BSA比较方式。对于抗病等这类具有两个差异很大的表型的数量性状,可以构建两个性状间具有质量差异的混池(图2a)。对于很多呈正态分布的性状,可分别选择极端高和极端低性状的个体构成混池(图2b)。为了增加统计学可靠性并且减少假阳性,可以在两个极端性状分别独立选择多个混池(图2c)。而在很多情况下,对于某些性状而言只有某一端的极端性状可用(比如另一端的极端性状个体被致死基因或严重的胁迫杀死),可以通过将一个混池与随机选择的正常个体进行比较来做BSA分析(图2d)。
有两种方式来将个体构成混池。可以将每个个体的组织混合在一起,然后提取一次核酸或蛋白;也可以分别提取每个个体的核酸/蛋白,然后等量混合。对于DNA分析来说,这两种混合方式并没有体现出很大的差异,所以在提取之间进行混样可能更加节约成本。
当只有一端的极端性状个体可用或者等位基因频率很难精确计算时,使用一端混池进行的BSA可以将该混池与随机选择的正常环境个体构成的混池进行比较,也可以与理论期望进行比较。一个类似的应用场景是目标性状与致死基因连锁,所以只有一端的个体能够存活,只能构建单个混池(图2d)。
为了增加BSA的定位能力,可以从同一个群体中构建多个平行的混池。只有那些真实的信号会在平行混池中都出现。这为我们寻找真实的遗传差异提供了确认方式,随着混池数量增加,假阳性出现的可能会下降。
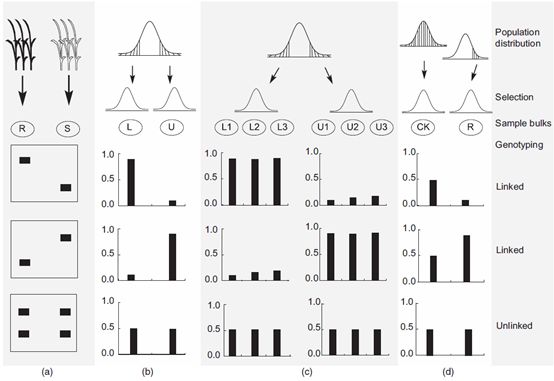
图2 BSA分析的4种类型
混合分析
可以对样本或标记进行混合分析。样本混合分析通常用于对个体进行选择的基因分型,可以将成本降到BSA的水平;而标记混合分析通常用于BSA当中。混合分析可以增加样本或标记的数量,但成本和时间并没有很大的增加。混池样本可以进一步进行混合分析,从而进一步降低成本,增加通量。
样本混合分析的一个典型例子是在每个样本测序时加上不同的识别序列(barcode),让多个样本同时上机测序,但数据分析的时候可以进行识别和拆分。可用的barcode的总数受到barcode长度和标签数量决定,在混样中使用双标签可以进一步增加可以混样的总数。在混样测序的情况下,一次单独上机可以对多个样本进行测序,降低成本,减少时间,提升效率。对于RNA来说,在BSA中分析使用的通常是其cDNA,所以DNA混合分析的方式也同样适用。
对于标记的混合分析来说,最早的尝试是在PCR分析中使用多对不同的引物。标记混合分析可以在DNA,RNA和蛋白等不同层面的研究中使用。在DNA或者cDNA层面,将上千个标记几种在一张芯片上可能是最佳的混合分析方法。在玉米当中已经开发了多种SNP芯片,SNP标记数量从1536-600 k不等。
混样测序可以在对DNA进行随机剪切后两侧连接上barcode并将所有样品混合上机测序,也可以通过限制性内切酶消化的方式来进行(比如RAD简化基因组)。前者可以用于快速对细胞器DNA和微生物DNA进行测序,也可以用于基因组SNP的挖掘和做图。后者通常用于高密度的SNP挖掘和基因分型。
分析:DNA,RNA与蛋白质
在新技术的加持下,两个差异样本之间的遗传差异可以使用分子标记、芯片或是高通量测序在DNA,RNA,或蛋白质层面进行。
DNA分析
在DNA层面,遗传差异可以通过DNA标记、芯片、简化基因组(比如GBS等)以及全基因组测序来进行挖掘。
传统的BSA方法通常使用DNA分子标记来进行,主要是基于PCR的分子标记。近年来基于芯片的基因分型方法逐渐开始流行,其优点是可以同时对大量的标记进行分型,通量很高,极高地提升了效率。
随着近年来测序成本不断下降,混池样本也可以通过不同深度的DNA测序来进行基因分型。测序产生的标记数量会随着测序深度的提升而增加,从而可以提升那些由多基因控制的复杂性状的定位能力。
全基因组测序可以用于性状分离个体的混合测序。分离群体可以通过突变个体与野生型亲本回交后自交产生,突变个体可以选择自然突变个体和化学诱变个体。使用突变个体构建混池的方法被称为Mutmap,使用突变产生的SNP作为标记来对含有目标突变的区域进行定位。这种方法已经成功地在水稻研究中得到应用。
RNA分析
通过RNA测序来进行基因分型有许多好处。首先,相对于芯片技术来说,通过RNA测序来进行基因分型可以检测出更多的突变。RNA通常可以覆盖所有基因的70%-90%,根据组织和发育阶段有所不同。比如测序70M reads(100bp)可以覆盖15日龄玉米种子的71.6%的基因。
RNA测序技术让我们可以让我们对特定物种的转录组进行解析,无论该物种有没有可用的参考基因组。与芯片等其他技术相比,RNA测序具备以下几种优势:首先,RNA测序并不要求物种特异或转录本特异的探针,所以可以对新转录本、基因融合、SNP、indel以及其他未知的变异进行探测。其次,与芯片杂交不同,RNA测序可以对基因表达的变化进行定量的探测。第三,RNA测序能够更灵敏,更特异地对基因、转录本和表达差异进行检测。第四,可以方便地增加测序深度,以检测稀有转录本或是微量表达的基因。除此之外,RNA测序还带来了更新现有基因注释,发现新外显子和连接位点的可能性。RNA-BSA可以在基因组中重复序列很多的时候显著地降低测序成本,例如玉米和小麦的基因组大小分别是2.4G和17G,其中前者的基因组包含高达80%的转座元件。
RNA测序的一个缺点是,当一个重要突变位于非表达的区域或者与可以被鉴定的SNP不连锁,就没有办法在作图过程中被挖掘到。另一个重要缺点是RNA测序无法对拷贝数进行检测,所以拷贝数变异也无法被加入到作图中。
单独的标记、蛋白质芯片、测序都可以用于鉴定蛋白质的种类和总量。在二维电泳中,蛋白质可以根据电荷量和分子质量被分离开。尽管此技术早在70年代便开始使用,比蛋白质组的概念要早很多,现在在许多实验室中这还是一个重要的研究手段,尤其在研究翻译后修饰方面。为了使用Wester blotting从复杂的蛋白质混合物中识别特定的蛋白质,蛋白质需要根据大小来分离,然后对目标蛋白使用抗体进行标记。
现在有3种蛋白质芯片,分析芯片(analytical microarray)、功能蛋白芯片(functional protein microarray)、反向蛋白芯片(reverse-phase protein microarray)。使用荧光标记的蛋白质芯片使用最广泛,也最灵敏。无标签(label-free)方法则提供了更多高通量蛋白质检测的可能性。
与DNA测序有多种平台可供选择不同,蛋白质测序目前只有两种主要的方法,即质谱(mass spectrometry)和Edman降解(Edman degredation)。由于灵敏度和效率关系,质谱不仅成为了研究蛋白一级结构的主要手段,也是蛋白质组研究的核心手段。当蛋白质被加上选择性标签(比如同位素标记亲和标签或者iTRAQ),就可以对部分蛋白质进行鉴定和定量。而当蛋白质被加上非选择性标签(同位素编码蛋白质标签或MCAT)时,可以对更大范围的蛋白进行鉴定和定量。如果蛋白质在具有代谢活性的时候加上标签(SILAC),还可以收集数千种蛋白质的动态信息。
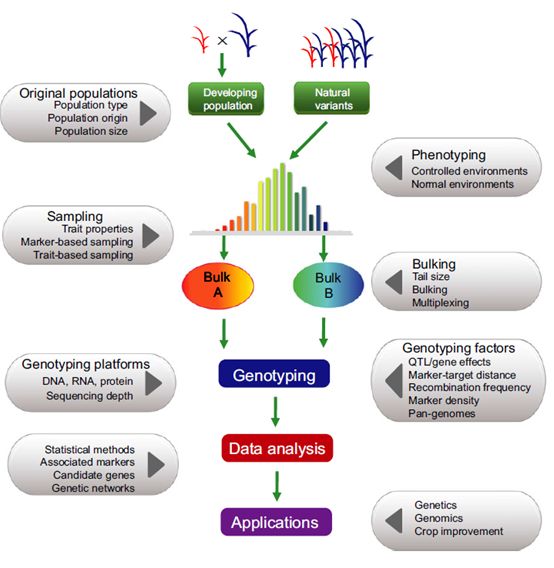
图3 BSA分析的流程与影响因素
应用:遗传学、基因组学和作物育种
遗传学
BSA在遗传学中的传统应用是遗传图谱和目标基因挖掘。最早的应用是使用分子标记和BSA分析来对重要农业性状的主效基因进行定位,比如水稻的产量、抗干旱、抗热,小麦的抗水分胁迫、埃及棉的抗盐等性状。
为了定位硫代葡萄糖苷积累的代谢QTL(mQTL),研究者在油菜DH做图群体中对硫代葡萄糖苷的总量和叶片与种子中的含量进行了分析。在两个器官中对硫代葡萄糖苷含量产生影响的QTL进行合并后一共有105个mQTL。在大米的研究中,研究者使用气质联用方式对两个亲本家系的糙米油中脂肪酸含量差别进行的鉴定,并且在F2群体中识别了29个相关的mQTL。mQTL作图同样在拟南芥中用于研究数量性状或质量性状,例如分析得到了22个与类黄酮化合物相关的QTL。
高通量基因分型手段的发展让BSA更多地使用芯片方式开展。这类方法已经成功用于研究玉米的隐性突变、小麦的锈病、拟南芥中硫和硒的含量、棉花的抗盐性状、大豆抗花叶病毒、辣椒抗腐根病以及白杨的光合作用性状等。
随着二代测序技术的发展,BSA可以通过对亲本和分离性状子代混池进行DNA或者RNA测序的方式来快速对候选基因进行定位。其中的一个典型例子是,研究者从一个10800个体的水稻F3群体中选择了430个极端不耐寒和385个极端耐寒的水稻秧苗,并且通过二代测序对450000个SNP进行了分型,找出了6个与耐寒性状相关的QTL,4个与抗稻瘟病相关的QTL,以及2个与秧苗活力相关的QTL。另一个典型例子是使用946个水稻F2个体将与水稻雄性育性基因ms-IR36定位到了染色体2短臂一段33kb的区域,区域包含10个候选基因。同时,高通量测序BSA还成功找出了黄瓜开花基因Flowering Locus T附近的早花QTL。高通量DNA测序BSA还用于研究棉花的短纤维突变。在部分研究中,这种方法还用于同时在单群体或多个平行混池中鉴定同一个基因。
使用二代测序和转录组从头组装的方式,可以通过RNA测序来进行类似的BSA分析和目的基因定位。在玉米中,这种方法已经用于进行突变基因的快速鉴定,使用32个突变个体和31个非突变个体对gl3位点进行了定位。另外的例子包括了小麦谷物蛋白含量位点的遗传图谱、小麦黄锈病的抗病性状、向日葵抗霜霉病性状、沙梨果皮色素沉积性状以及萝卜的细胞质雄性不育等。
基因组学
功能分析
高通量的方式极大促进了功能基因组学的发展,包括转录组和蛋白组学。
蛋白芯片在人和动物研究中有五种主要的应用,包括疾病诊断、蛋白组学、蛋白功能分析、抗体鉴定和新疗法开发。例如,基于BSA的功能基因组学鉴定出了在成人卵母细胞中mRNA丰度更高的193个基因和青春期前卵母细胞中mRNA丰度更高的223个基因。
在鼠李糖乳杆菌(Lactobacillus rhamnosus)中,100株不同来源的菌株被用于通过基因组与表型的关联分析来研究基因组复杂性以及生态学功能。将这些菌株的基因组与参考基因组序列比对,对一系列代谢、抗性、功能性状进行了鉴定。
在植物中,蛋白功能分析是基因组学研究的一个主要应用,它可以被用于鉴别蛋白-蛋白互作、蛋白-磷脂互作、酶反应底物以及受体配体等。
随着液质联用方法的快速发展,大范围的蛋白质组鉴定和定量技术逐渐成熟。在植物中,有通过蛋白质组对可能具有农业应用价值的蛋白进行检测的研究。在小麦面粉中使用质谱鉴定出了200种醇溶蛋白和谷蛋白,并对丰度进行了鉴定。在大豆中鉴定出了4975种核蛋白,并且推断在易感锈病和抗锈病个体中差异表达的蛋白可能与抗性性状存在关系。使用二维电泳加液质联用的方法,在拟南芥中鉴定出了42个受油菜素甾醇(brassinosteroid)调控的蛋白。这些蛋白预计在特定的细胞过程中扮演重要角色。
表型的表现通常呈现环境依赖的关系。环境可以被定义为对生物体产生影响的因素的总和。农作物在不同的环境栽种时,面临的土壤类型、肥沃程度、含水量、气温、光周期、生物/非生物胁迫等都不同,其基因的表达也可能相应地受到调控以对这些内外因素做出反应。一个特定的基因型或品种的作物在不同的环境中表型变化可能会呈现一个特定的范围,即为表型的可变性。环境互作基因分型通常将研究重点放在个体的表型性状上,其中三种主要的研究方法,包括使用双亲本群体绘制遗传连锁图、自然群体GWAS和遗传图谱-GWAS综合分析。将GWAS与转录网络和蛋白成分表型结合可以对候选基因进行挖掘和验证。将生理生态模型与遗传作图方法相结合来构建基于QTL的模型可以将表型和基因型的研究联系起来。例如基因的表达可以通过对不同的个体在不同的发育阶段以及不同的环境下进行全基因组测序并且与表型进行关联。
在这种方法中,全基因组分析策略拥有很强的能力,在基因组、环境、空间和时间4个维度上的变化可以让不同条件下基因表达的变化清晰地呈现出来。为了将这种分析落到个体层面,需要检测个体中特定基因的单倍型,从而推断出受到环境影响的性状相关联的QTL。
这种通过环境互作进行基因分型的分析方法可以通过良好的环境管理来进一步加强准确性,但是,从受控的温室中得出的结论并不能直接推广到田地环境中。拟南芥中的数个研究支持了这一论点,拟南芥在温室中的开花时间和田地中的开花时间关联非常微弱。
将最新的测序结束和BSA分析相结合可以得到快速鉴定基因和突变的有效工具,可以用在育种的标记开发过程中。相较于对整个群体进行研究的策略,BSA可以提供开发重点性状标记的捷径。除了常用的DNA分子标记,RNA和蛋白质分析也同样可以用于标记的开发。为了更有效地根据标记选择个体,标记的选择应当从基因及其功能出发。为了揭示基因内部的等位基因变化带来的影响,可以使用多个标记来构建单倍型,以呈现不同等位基因之间的相互组合。在一个特定区域内,标记密度越大,构建的单倍型就越有研究意义。
随着RNA测序技术的发展,农业技术措施对植物生长发育的影响可以通过对不同措施下植物基因表达的变化进行测定来进行研究。农业基因组学可以指针对目标性状的基因表达受到农业技术措施影响的研究,通过这些研究我们可以开发高效、低成本、环境友好的措施来增进作物产量。我们可以直接研究环境中影响植物基因表达的因素,但是,在研究农业措施对基因表达的影响的同时,复杂多变的环境因素也会产生很强的干扰。农业基因组学可以更好地解决育种中的这些问题。
其中一个例子,从稻田中收集的稻叶的转录组数据和对应的气象数据被用于研究自然环境下水稻转录组的变化,并构建了影响基因表达因素的统计模型。该模型使用了不同取样时间的461个芯片数据以及对应的风速、气温、降水、全球太阳辐射、相对湿度以及气压等气象数据。 模型的可靠性分别使用栽种季节的田地作物与受控条件下温室作物的芯片数据进行了验证。昼夜节律、气温、植株年龄和太阳辐照度是控制转录变化的最主要因素,从而验证了根据环境因素对作物的相关生物学过程产生影响的预测方法。
标记辅助选择与选择性基因分型
为了更有效地依据标记进行育种选择,一个大的群体可以根据标记-性状之间的联系或群体特征分成数个亚群体,并在亚群体中进行BSA分析来提升选择的效率,还可以在亚群体中使用选择性基因分型的方式来确认满足需求的个体。
选择性基因分型,即对目标群体中部分个体进行基因分型,是在目标基因区域、等位基因或单倍型已经得到充分研究时可采用的有效方法,尤其是研究那些难以测定或成本很高的性状时。在测序和基因分型的成本迅速降低的条件下,我们可以首先对整个群体进行基因分型,挑选信息量最丰富的个体组成亚群体,并对亚群体中的个体进行精确的表型鉴定。与不加选择的表型鉴定相比,选择性分析在对同样数量的个体进行表型鉴定的情况下,可以获得更多变异信息,具备更强的定位能力。对于表型鉴定来说,鉴定那些极端表型的个体通常更加容易,例如研究非生物胁迫时,大量个体会直接被强烈的胁迫淘汰。
在植物发育的早期阶段进行高密度的栽培与选择可以在同等条件下研究更多的植株个体,是在对某些性状进行研究时可以采用的方案。在研究一些受到种植密度强烈影响的性状时这种方法可能会带来很大干扰,但是在研究一些受干扰很小的性状时可以考虑采用这种方法。
展望
BSA方法具备前文所述的种种优点,但同时也有一些不可忽略的缺陷。尽管BSA已经在基因组学和遗传学上有诸多应用,但是它还是主要研究那些受主效基因控制的性状。对于那些受不同效果基因控制的复杂的数量性状而言,混池样本的选择可以通过环境互作进行表型鉴定的方式来进行,这样那些相对来说表型贡献度更大的基因通常可以被定位到。但是对于那些受很多微效基因控制并且受环境显著影响的复杂性状,BSA的分析能力可能显得不足,选择到那些包含目的等位基因的极端性状个体的可能性会变得很小。与那些传统的遗传学分析方式相比,BSA仅考虑那些呈现出极端性状的个体,而忽略其他个体的性状信息。其结果就是,BSA分析中表型方差、LOD值、加性效应等分析会出现偏差。并且,BSA对上位效应分析无能为力,而且对表型鉴定的偶然差错非常不敏感。不过,BSA可以通过增大群体大小、极端性状混池大小以及标记密度等方式进行改进,也可以使用多个平行混池、更加精细的表型鉴定和环境控制来提升分析能力。除此之外,还可以通过GWAS来对BSA的结果进行验证,并对一些复杂的结果进行进一步解析。
随着测序成本的显著下降,尤其是RNA测序技术的成熟,基因分型变得更加方便快捷。这也使得BSA可以在一个更大的群体中对多个混池进行分析,从而提升检测能力,可以检出那些稀有突变或等位基因。当确认BSA测序的策略时,需要仔细考虑混池个数、混池中的个体数以及测序深度等因素。
与DNA芯片相比,蛋白质芯片的开发更加麻烦且困难。技术上的困难是找到一个让蛋白质附着和固定的平面,并且让蛋白质能够保持二级结构或三级结构,并且维持足够的时间不被降解。实验上的困难是获得能够结合目标基因组中所有蛋白质的抗体或其他结合分子、对结合的蛋白进行定量、从芯片上提取目标蛋白进行后续分析以及减少非特异结合等。最后,蛋白芯片还需要提升呈现整个蛋白质组,减少高丰度蛋白影响的能力。
从技术上来说,基于芯片的蛋白互作高通量分析以及其他生物分子的互作拥有非常广阔的前景,可以完整呈现出各种信号通路和网络。随着蛋白质组技术分析通量的极大提升,在一天之内完成对生物体各个器官组织的比较逐渐成为可能。
尽管BSA在DNA和RNA层面得到了广泛应用,在蛋白层面进行的BSA目前还未有报道。鉴于许多基于质谱的蛋白定量方式可以对两个样本进行比较,我们可以期待其中的一些方式也可以应用于BSA-蛋白分析。基于蛋白的BSA的一个主要的困难是纤维素的富集以及许多植物的次级代谢产物,使得从组织中提取蛋白的过程非常困难。
传统的基因分型方式,例如单独的分子标记或者芯片,可以通过测序的方式进行补充,包括常规的全基因组测序以及简化基因组测序。简化基因组技术可以降低基因组复杂性,提升灵活性和效率,得到足够的标记数量,同时成本相对较低。简化基因组可以用于GWAS、遗传多样性分析、遗传连锁分析、分子标记挖掘以及作物育种选择上。当两个混池中个体数足够多(比如超过500)的时候,BSA可以与简化基因组联合使用。
可以预见,已经在遗传图谱和基因定位方面取得成功的BSA会在遗传学、基因组学、作物育种等方面拥有越来越高的重要性,并且会在某些方面逐渐取代对整个群体进行基因分型的研究方式。通过高通量测序来进行的BSA会随着新分析软件、新的单倍型鉴定方法、新的低频变异挖掘技术以及新的读长更长的测序技术不断发展而变得越来越具吸引力。
- 本文固定链接: https://maimengkong.com/zu/1220.html
- 转载请注明: : 萌小白 2022年10月3日 于 卖萌控的博客 发表
- 百度已收录
