大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
染色质免疫共沉淀(Chromatin Immunoprecipitation,ChIP),是一种用来在体内鉴定及分析基因组内部蛋白质-DNA 相互作用的有力研究技术。在ChIP的基础上,用二代测序检测ChIP实验的DNA产物,将ChIP与高通量测序技术相结合的ChIP-seq技术,可在全基因组范围对特定蛋白的DNA结合位点进行高效而准确的筛选与鉴定,为研究的深入开展打下基础。
本期易基因小编为您介绍ChIP-seq的主要研究方向、研究思路(前期探索性实验、数据挖掘思路、下游实验设计)、并聚焦ChIP-seq实验成功的关键问题,一起来看看吧。
一、CHIP-seq的主要研究方向
ChIP-seq技术能够提供更高的分辨率、更少的噪音和更大的覆盖范围,然而其只能在明确感兴趣的蛋白(或转录因子)后,才能针对性富集相应的DNA,因此,ChIP-seq需要与其他技术和方法相配合才能更好的进行表观基因组的研究。
(1)顺式调控元件
- PromoterEnhancer/Super enhancerSilencerOperator
(2)反式作用因子
- 转录因子转录辅因子阻遏子
二、CHIP-seq的研究思路之前期探索性实验
1、充分的文献和数据库调研
- DNA与蛋白互作是研究得最早的表观遗传修饰机制之一,许多领域都有大量 的研究积累,数据库中也集成了大量的数据,根据选题充分地进行调研很重要。根据已有知识提出自己合理的科学问题和假设。图谱型研究最好要结合转录组测序。参与感兴趣生物学过程的重要的转录因子有哪些?相关研究对象有没有增强子/超级增强子、转录因子数据库可以用?(ENCODE,modENCODE等)基于已有的研究机制,有哪些不足?还能够做哪些深入研究?
2、转录因子/辅因子/组蛋白修饰基因的干扰/敲除/过表达
- 检测处理后目标转录因子/辅因子/组蛋白修饰基因的转录和蛋白表达检测处理后研究对象的表型和功能变化
eg:根据文献调研,推测转录因子Irf8可能在第三类骨细胞分化形成过程中起关键作用,于是首先对Irf8进行敲除实验,然后通过对比处理前后骨细胞分化的情况来确定该转录因子的作用。
3、其他前期实验
在没有条件开展敲除/过表达实验的情况下,也可以开展其他预实验。
- 关键表型指标测定(比如不同发育阶段的表型差异,不用任何处理就能检测到差异)转录组(成本低廉)—— 基于DNA与蛋白互作会影响基因转录
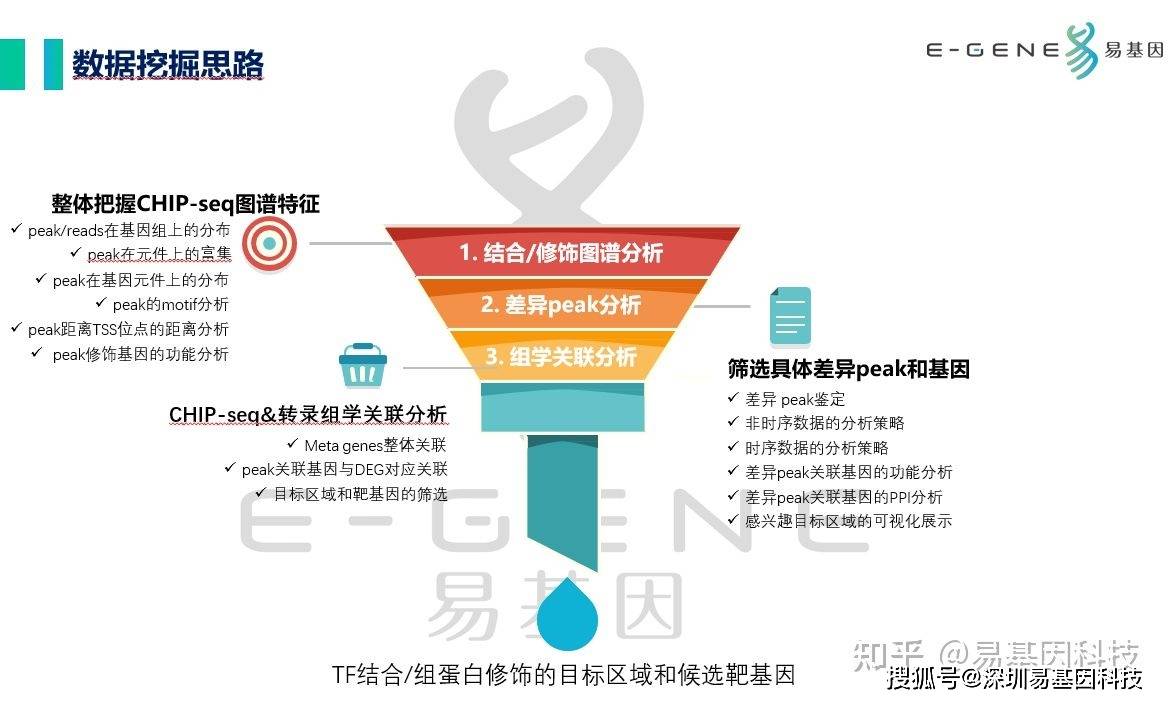
三、CHIP-seq的研究思路之数据挖掘思路
1、图谱分析
(1)peak/reads在基因组上的分布
- Peak的分布就是蛋白与DNA互作图谱。不同蛋白对DNA的结合可以按照峰的宽窄和分布特征分为:narrow peak:即发生在DNA上特定的短序列,结合的区域很短。broad peak:这种类型的peak在DNA上呈弥 散的连续的分布,峰型较宽。一般来说,转录因子的峰型都是narrow peak;而对于组蛋白修饰,有的峰型为 narrow peak,有的为broad peak。可以通过调整参数或使用不同的软件分别鉴定narrow peak及broad peak。
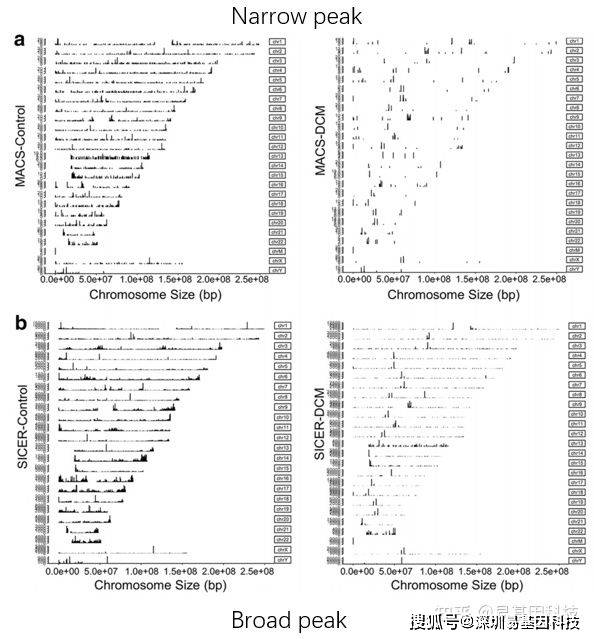
peak分布圈图
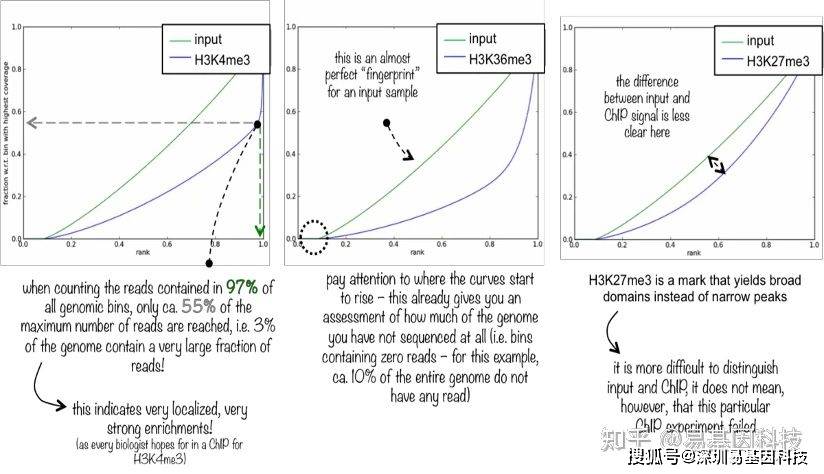
(2)信号的富集程度分析——覆盖度累积曲线
对样本比对结果reads累积情况进行展示。一定长度窗口(bin)上reads数进行计数,然后排序,再依次累加画图。input (能测到90 DNA片段)在基因组理论上是均匀分布,随着测序深度增加趋近于直线,实验组在排序越高的窗口处reads累积速度越快,说明这些区域富集的越特异。narrow peak :富集程度高;broad peak:富集程度低。
- 富集程度低不代表失败, 如broad peak。但是如果是转录因子, 富集程度低则需要谨慎对待。
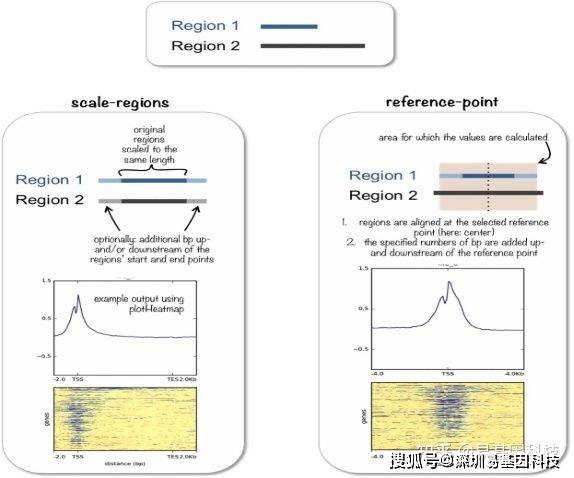
(3)peak/reads的基因元件富集分析
- reference-point(relative to a point): 计算某个点的信号丰度scale-regions(over a set of regions): 把所有基因组区段缩放至同样大小,然后计算其信号丰度。
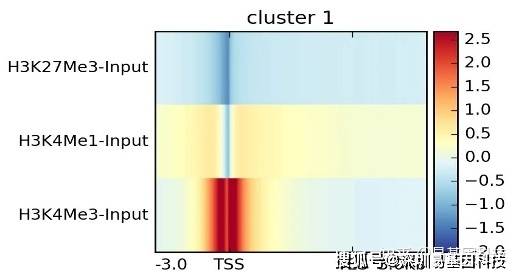
- 基于信号富集的靶基因集分类鉴定(基于聚类算法)
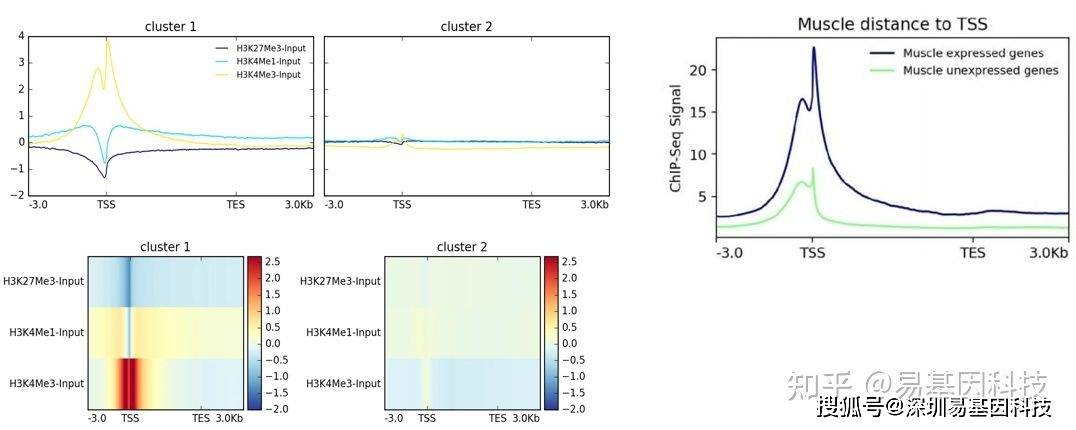
(4)peak/reads的基因元件分布分析
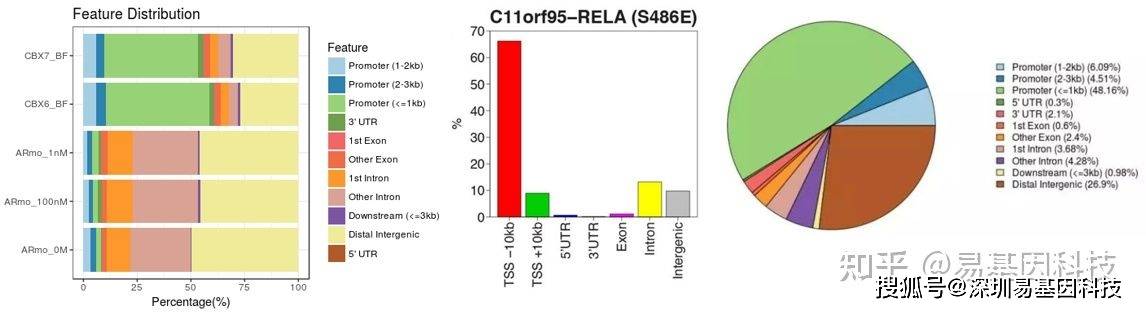
(5)peak/reads与TSS的相对距离分布
转录因子、组蛋白修饰往往具有重要的转录调控功能,而TSS附近是主要的转录调控区域,因此判断peak与TSS的位置关系有重要的意义。
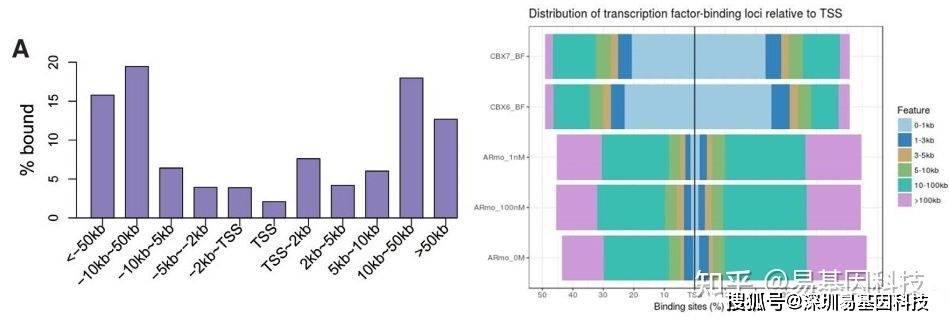
(6)降维分析
将基因组分为等长窗口(bins),计算各样本各窗口内的Reads覆盖情况并进行标准化。基于此数据进行相关性、聚类和PCA分析。

(7)motif分析
Motif为一段有特征的DNA短序列,主要为转录因子的识别位点,不同的motif对应不同的转录因子。
- 根据motif可以推测结合的转录因子。已知转录因子则分析该转录因子识别的序列特征。

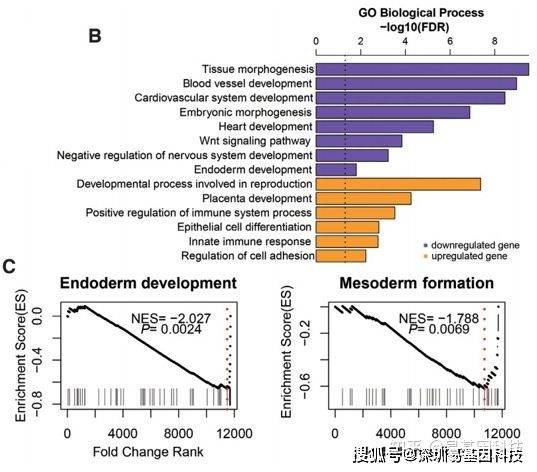
(8)peak的基因注释和功能分析
- ORAGSEA: 可以按照peak信号强度排序
2、差异peak分析
(1)非时间序列数据:

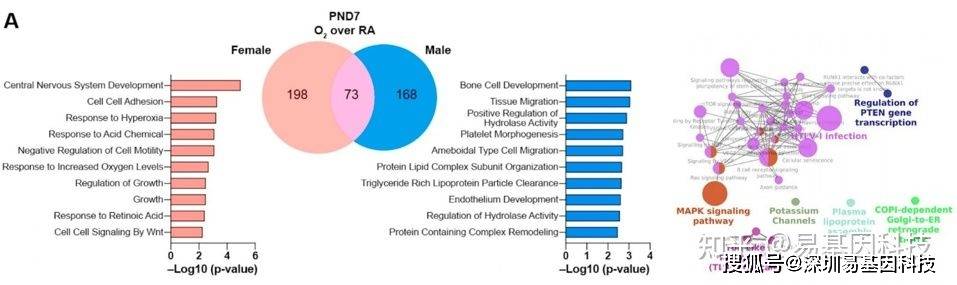
(2)时间序列数据:

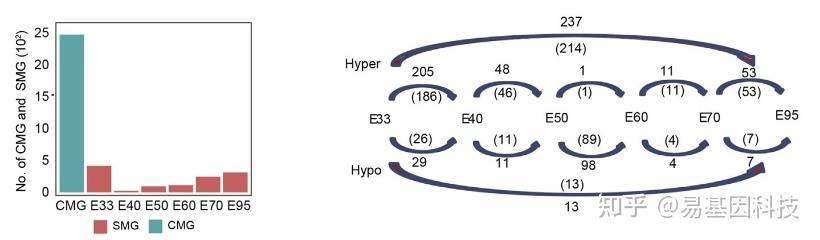
(3)差异peak关联基因的PPI分析

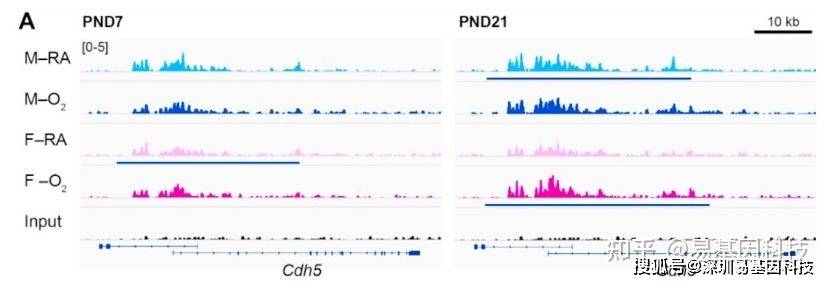
(4)感兴趣基因的差异peak展示
3、组学关联分析:CHIP-seq&转录组学
(1)Meta genes整体关联
- 距离TSS位点不同距离的peak注释到的基因的表达水平分析不同表达水平的基因,peak的数量分布对比
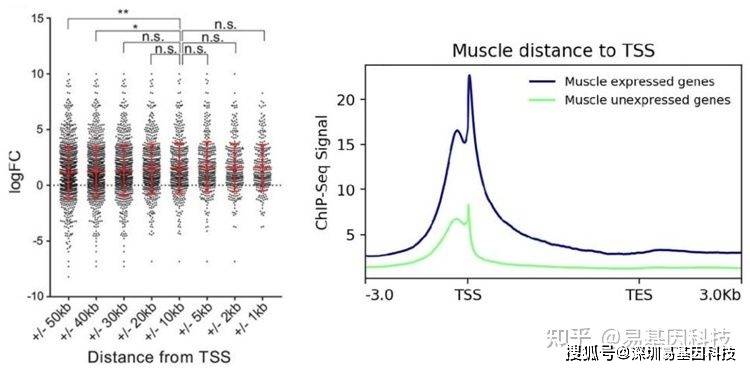
转录水平倍数变化 vs. peak倍数变化
(2)差异peak基因-DEG对应关联:筛选关键目的基因:
- peak关联基因与差异表达基因的重叠分析。peak关联基因可以是peak注释到启动子区,TSS±10kb区的基因,也可以来自已 知公共数据库的注释,如Human Enhancer Disease Database (HEDD)。九象限图法
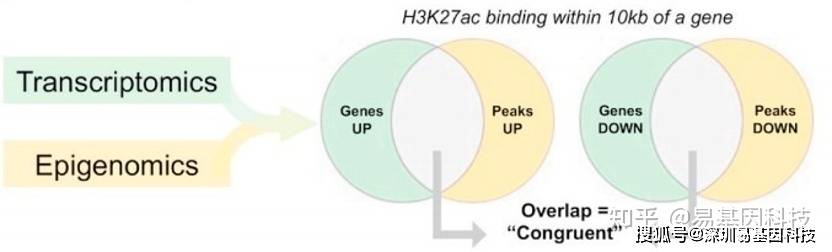
4、CHIP-seq数据挖掘思路
(1)整体把握CHIP-seq图谱特征
- peak/reads在基因组上的分布peak在元件上的富集peak在基因元件上的分布peak的motif分析peak距离TSS位点的距离分析peak修饰基因的功能分析
(2)筛选具体差异peak和基因
- 差异 peak鉴定非时序数据的分析策略时序数据的分析策略差异peak关联基因的功能分析差异peak关联基因的PPI分析感兴趣目标区域的可视化展示
(3)CHIP-seq&转录组学关联分析
- Meta genes整体关联peak关联基因与DEG对应关联目标区域和靶基因的筛选
四、CHIP-seq的研究思路之下游实验设计
1、简单验证
(1)目标基区域转录因子结合/组蛋白修饰的验证:CHIP-qPCR
(2)检测目标基因的mRNA表达水平:RT-qPCR
(3)检测目标基因蛋白质表达水平:Western blot
2、转录因子或组蛋白修饰添加基因的干扰实验(非靶向),验证是否影响目标基因表达和细胞功能
(1)转录因子或组蛋白修饰添加基因的干扰:基因的突变/敲降/敲除/过表达、相关蛋白的抑制剂
(2)检测TF结合/组蛋白修饰的整体变化:CHIP-seq
(3)检测目标区域的TF结合/组蛋白修饰的变化:CHIP-qPCR
(4)检测下游靶基因的mRNA水平:RT-qPCR
(5)检测下游靶基因蛋白质水平:Western blot
(6)检测细胞功能/表型变化
3、靶向目标区域的TF结合/组蛋白修饰干扰实验,检测其影响下游靶基因的表达
(1)目的基因结合/修饰干扰细胞系的构建:荧光素酶活性分析实验(Luciferase activity assay)、CRISPER-CAS9靶向目标区域引入突变/修饰
- 确定并验证与转录因子结合的motif验证与该motif结合之后是否能影响靶基因表达不同区域引入突变以确定关键结合位点
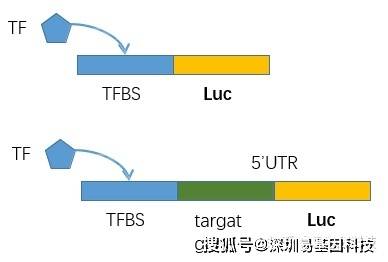
用于验证TF结合并调控下游基因的荧光素酶报告系统
(2)检测目标区域的TF结合/组蛋白修饰变化:CHIP-qPCR
(3)检测荧光素酶报告基因的mRNA表达水平:RT-qPCR
(4)检测目标基因蛋白质表达水平:Western blot
(5)检测细胞功能受到的影响:免疫荧光显微观察、功能标志物测定
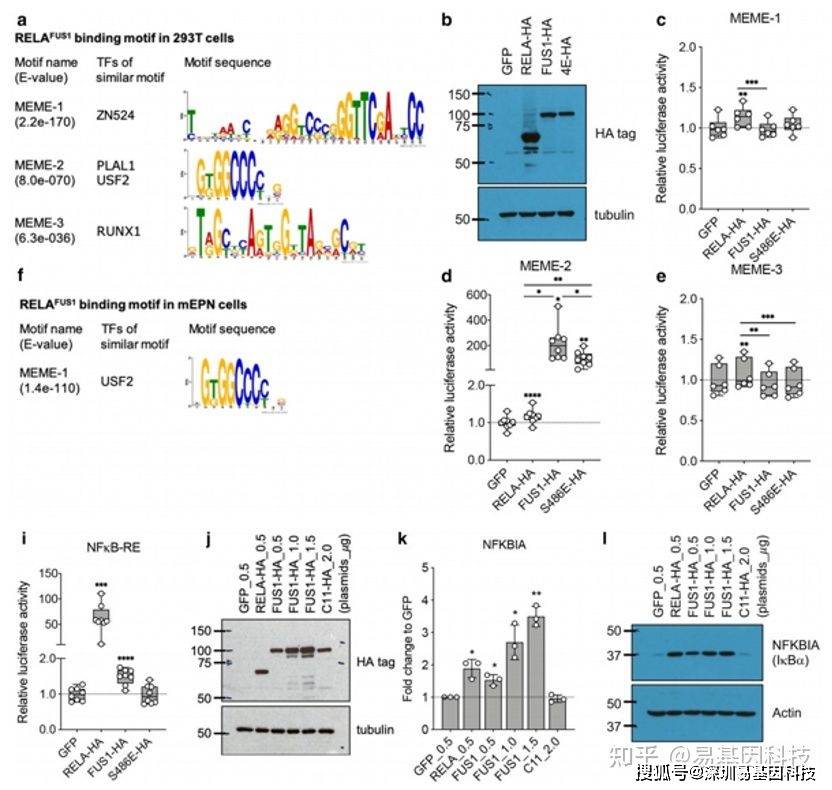
4、荧光素酶实验验证转录因子结合的motif,并证明结合后直接调控靶基因的表达
top3的motif中,只有MEME-2能够激活荧光素酶基因表达,表明FUS1转录因子结合的是MEME-2 motif。
在靶基因3’UTR插入荧光素酶基因,只有FUS1组能够激活荧光素酶基因的表达,表明只有FUS1组具有转录因子活性。
top3的motif中,只有MEME-2能够激活荧光素酶基因表达,表明FUS1转录因子结合的是MEME-2 motif。
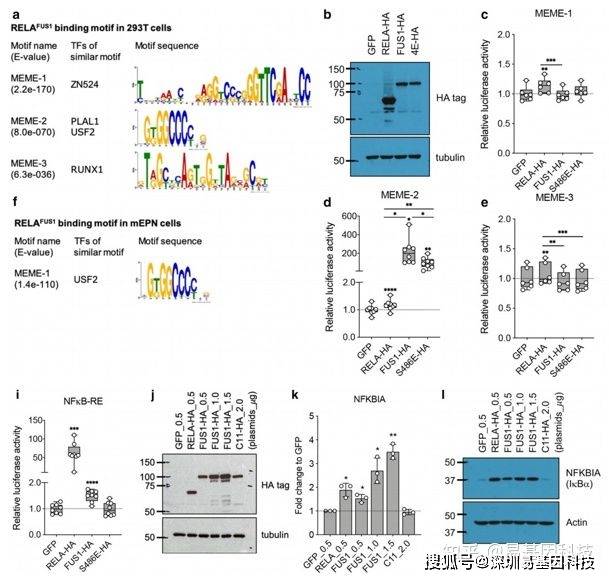
引入S486E突变影响到该转录因子对MEME-2的结合,也最终影响到靶基因的表达。
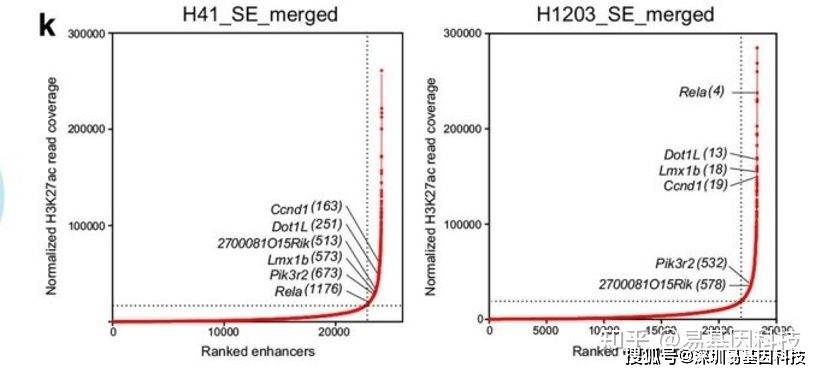
5、超级增强子
- 根据通用型转录辅因子Med1在CHIP-seq实验中富集程度的高低,可以将增强子分为以下两类:typical enhancers(TE):长度为几百bpsuper enhancers(SE):长度几千bp超级增强子的预测建立在增强子的基础上,可以看做增强子富集的区域。相比增强子,超级增强子区域具有更高的转录因子的密度,能够让转录效率提高达几百倍。H3K27ac是活性增强子的标志,可以通过H3K27ac CHIP-seq鉴定TE和SE。
五、CHIP-seq实验成功的关键问题
(1)抗体质量
ChIP-seq是基于抗体的免疫沉淀实验,因此它的数据质量好坏直接取决于抗体的质量和特异性。
另外,针对同一蛋白的不同抗体,可能会识别不同的表位(尤其是单克隆抗体)。因此建议针对同一感兴趣蛋白测试不同的抗体,通过Western blot检测knock-down前后的差异帮助选择。
(2)测序数据量
为了捕获所有真实的结合位点,而我们看不见摸不着,只能通过测序的reads去计算来帮助判断,因此测序reads的数量是一个决定因素。
需要多少reads呢?
这个取决于基因组的大小和感兴趣因子的结合方式(sharp regions for TFs and broad regions for histone marks)。哺乳动物中,鉴定TFs至少要满足10-20M,broad histone marks至少要10-45M,input对照要和ChIP样本保持同样测序深度。reads数量还取决于抗体质量和免疫沉淀的效率。信噪比越高,需要的reads数可以适当减少。
(3)生物学重复
样本重复可以看到实验设计的好坏,选择相关性高的样本进行后续分析
推荐三个生物重复,但两个现在也能接受(最粗略的实验设计就是:每个ChIP样本2个重复,input只有一个没有重复)
如果样本间的本质差异越大,越需要设置重复,例如从不同人取的样本。
关于染色质免疫共沉淀测序 (ChIP-seq)
染色质免疫共沉淀(Chromatin Immunoprecipitation,ChIP),是研究体内蛋白质与DNA相互作用的经典方法。将ChIP与高通量测序技术相结合的ChIP-Seq技术,可在全基因组范围对特定蛋白的DNA结合位点进行高效而准确的筛选与鉴定,为研究的深入开展打下基础。
DNA与蛋白质的相互作用与基因的转录、染色质的空间构型和构象密切相关。运用组蛋白特定修饰的特异性抗体或DNA结合蛋白或转录因子特异性抗体富集与其结合的DNA片段,并进行纯化和文库构建,然后进行高通量测序,通过将获得的数据与参考基因组精确比对,研究人员可获得全基因组范围内某种修饰类型的特定组蛋白或转录因子与基因组DNA序列之间的关系,也可对多个样品进行差异比较。
应用方向:
ChIP 用来在空间上和时间上不同蛋白沿基因或基因组定位
- 转录因子和辅因子结合作用复制因子和 DNA 修复蛋白组蛋白修饰和变异组蛋白
技术优势:
- 物种范围广:细胞、动物组织、植物组织、细菌微生物多物种富集经验;微量建库:只需5ng以上免疫沉淀后的DNA,即可展开测序分析;方案灵活:根据不同的项目需求,选择不同的组蛋白修饰特异性抗体。
技术路线:
- 本文固定链接: https://maimengkong.com/zu/1192.html
- 转载请注明: : 萌小白 2022年9月30日 于 卖萌控的博客 发表
- 百度已收录
