毫无疑问多组学联合的实验设计对于解析生物学问题是十分有效且敏感的。相对于单组学来说,起到的是1+1>2的作用。多组学就像是一条河流,从最初的DNA(基因组)到转录水平(转录组)以及解析来进行翻译(蛋白组),以及下游对于代谢物质的研究(代谢组)。同时还有多种表观修饰发挥作用(表观组学),不同的外部微生物环境也会造成差异(微生物组)。从变异或者表达水平单方面去解析生物学问题,毫无疑问在当今的研究中是较为乏力的。多组学可以从多个方面来解析问题,并且多种组学可以互相验证,在挖掘调控网络、研究分子功能都可以起到很大的作用。
对于当今火热的单细胞领域来说,除了单细胞组学间的互相联合分析(如scATAC、scRNA联合分析;scRNA、空转联合分析)之外,结合单细胞和其他组学研究能碰撞出怎样的火花呢?今天给大家分享一篇结合了scRNA、scATAC、EQTL(基因组、转录组)等组学的研究文章。
2021年,宾夕法尼亚大学肾脏电解质和高血压科等基构在《Nature Genetics》上发表论文《Mapping the genetic architecture of human traits to cell types in the kidney identifies mechanisms of disease and potential treatments》。
开篇明义,作者介绍到慢性肾病(CKD)影响着全球八亿多人,全球每60人死亡其中就有1人患有肾病。所以了解慢性肾病遗传学的特征是十分关键的。当前的研究中,基于大队列的GWAS发现了约300个位点与肾功能具有统计学意义的相关性。然而,解析这些位点的功能影响还是十分困难的。首先,目前尚不清楚这些变异在哪种细胞类型中是活跃的,以及它们如何影响特定的生物途径。另外就是超过90%的主要相关变异位于基因组的非编码区,使CKD风险靶基因的精确识别更加困难。
当前将定位变异到致病基因的常用方法就是利用eqtl,同时结合表观遗传学的方法来分析重要的基因功能调控区域。作者在这篇文章中,结合了多种单细胞测序方法,将细胞类型,变异,基因功能进行了整合分析以试图解析复杂的肾脏疾病相关表型。
接下来我们来看下作者的分析方法与主要结论。
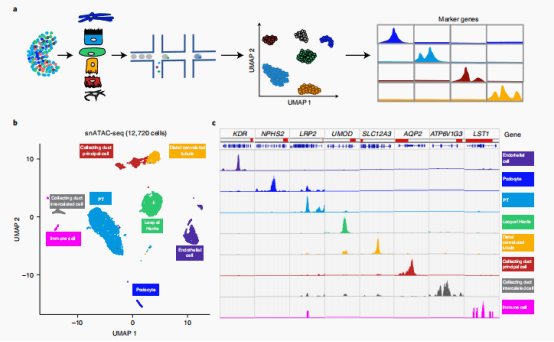
一:利用scRNA-seq解析人肾样本的细胞分数,并且鉴定eQTL(cf) eGenes
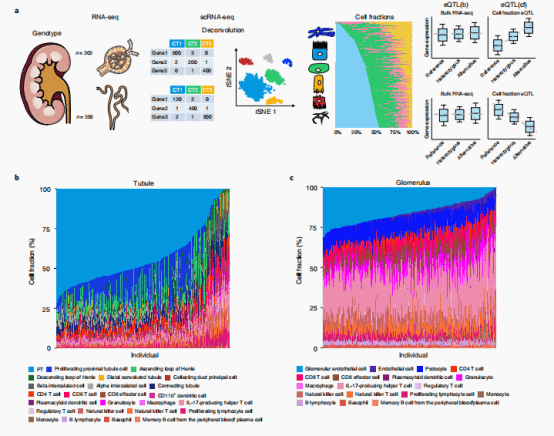
作者一共收集了659个样本,为了减少细胞的异质性,作者将每个组织样本显微解剖到肾小球和小管隔间,对这并659个样本(356个小管和303个肾小球)进行rna测序和基因分型。基于常规的EQTL分析,作者在小管和肾小球样本中分别发现了3599和5871个eGenes(该基因与具有显著P值的SNP相关)。
接下来作者提出解析每个样本的细胞群分布可以提高eQTL信息的质量,作者首先对但肾单细胞数据集进行分析并且定义出细胞类型,基于这些细胞类型与表达对bulk RNA-seq进行反卷积(CIBERSORTx方法)分析。与预期的一样,近端小管(PT)细胞是显微解剖小管腔室中最常见的细胞类型,肾小球内皮细胞是肾小球细胞主要细胞类型。作者也注意到PT细胞分数与估计的免疫细胞分数和肾纤维化呈负相关。加入细胞分数(cell fraction)之后,eGenes的数量在肾小管和肾小球中分别增加了28%(1256个基因)和18%(1194个基因)。
使用eQTL细胞分数的eQTL(cf)模型,即将样本细胞分数作为协变量因子,通过表达式残差(PEER)因子的概率估计来估计潜在变量的调整。该分析分别在小管和肾小球样本中鉴定出9209和10106个egenes,其中6821和7501个蛋白编码基因,865410和897548个显著snp基因对。
作者发现基于eQTL(cf) 模型发现的egenes相较于之前具有两倍的数量。一些之前没有发现的基因,如SANBR/KIAA1841和MRPL43在之前的eQTL分析中没有显著相关性,但在eQTL(cf)模型中却显示出了显著的相关性。并且该模型发掘的egenes与之前报道的SNP-GENES配对也有很好的重复性。
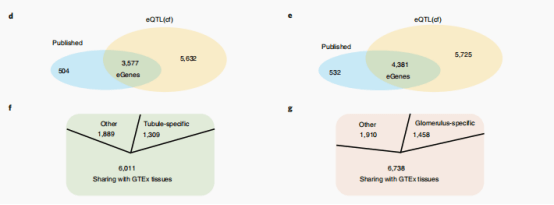
作者发现在625个(编码功能缺失)肾病基因中,有303个被鉴定为eGenes。GO分析表明,eGenes在代谢过程中富集,包括小分子和碳水化合物衍生物的代谢过程。对发表的GTEx数据中48个组织的meta分析显示,与eQTL(cf)研究中存在大量的共享基因,同时还包括1309个小管特异性和1458个肾小球特异性egenes(M>0.9)。
二、接下来既然已经知道每个样本的细胞成分,那么作者就关注到那些与细胞类型相关的eqtl
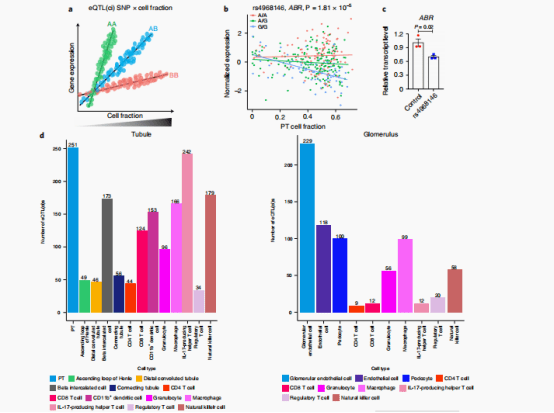
细胞类型相互作用的eQTL(eQTL(ci)),以细胞类型依赖的方式调节基因表达,作者进行了线性回归分析,模拟了细胞分数和基因型之间的相互作用。
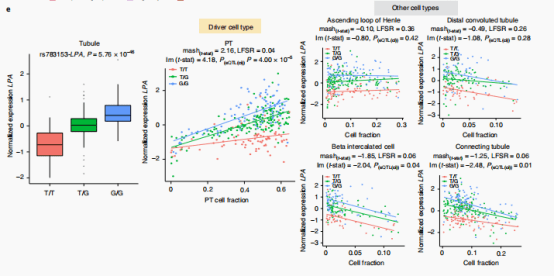
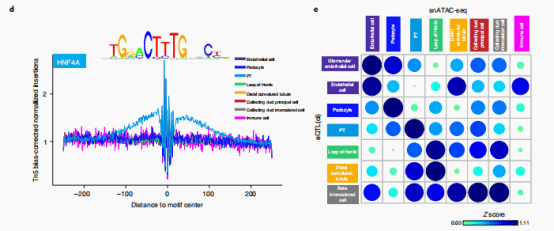
作者在肾小管和肾小球样本中分别鉴定出1613个和713个蛋白编码eQTL(ci)基因。接下来作者举了些例子,例如在G等位基因剂量较高(SNP rs4968146)的个体的肾脏中,PT细胞组分与ABR表达呈负相关,表明细胞类型依赖的基因型-基因表达相互作用。明确rs4968146所在基因组区域在调节ABR表达中的作用。作者在培养的人胚胎肾细胞(HEK293)中进行了基于CRISPR的基因组编辑验证。在该区域缺失后,ABR的表达明显降低。除此之外,作者还使用了multivariate adaptive shrinkage method (mash)方法定义了特异细胞类型对应的eQTL(ci)(具体见下图D)。接下来作者也具有更多的例子,具体参见图E。
三、作者接下来结合了snATAC-seq构建了单细胞分辨率的调控图谱
snATAC-seq分析确定了359,019个可及性染色质峰。并且作者基于不同亚群之间的peaks差异分析,鉴定出了60,661个细胞类型特异性peaks。为了确定这些细胞类型特异性开放染色质区域的转录因子,作者使用chromVAR和homer进行了motif 富集。分析发现HNF4A基序在PTs中富集,ESRRB在髓袢中富集,ERG在内皮细胞中富集。通过对8种主要细胞类型的转录因子足迹分析显示,在PT细胞中,HNF4A和HNF4G特异性富集。作者发现,eQTL(ci) 相关的 SNP 通常被预测为会破坏特异细胞类型转录因子的结合基序;其中一个例子是rs12481710-SYCP2对,它被预测会破坏PT细胞特异性转录因子HNF1A的结合基序。
接下来,作者估计了eQTL(ci)变异在相关的细胞类型特异性开放染色质区域的富集情况。其中发现丰富的肾细胞类型(肾小球内皮细胞、内皮细胞、肾小囊脏层细胞、PT细胞、髓袢细胞和Beta intercalated细胞) ,eQTL(ci) snp在相应细胞类型的开放染色质区域的富集,表明了细胞类型互作的eqtl与相应的细胞类型特异性调控区域之间的一致性。同时,eQTL(cf)的变异虽然也在snATAC-seq中也存在明显的富集,但是并没有细胞特异性。
同时作者利用贝叶斯共定位检验整合6个肾脏相关性状和eQTL(cf)数据,作者在肾小管中发现了240个共定位蛋白编码基因,在肾小球室中发现了230个基因。其中FGF5作为SBP(收缩压)和eGFR(估算肾小球滤过率)的靶基因,在SNP rs3822015处的C等位基因剂量与肾小管中FGF5的高表达有关。SBP和eGFR位点±100kb内的snp的eQTL和GWAS效应大小之间有很强的相关性。作者将与SBP、eGFR相关的SNPs比到PT 特异的开放染色质区域,发现rs3822015具有显著性,该SNP也破坏了一个假定的HNF4A转录因子结合的motif位点。并且作者使用CRISPR进行了验证敲除该区域会明显影响FGF5的表达水平。
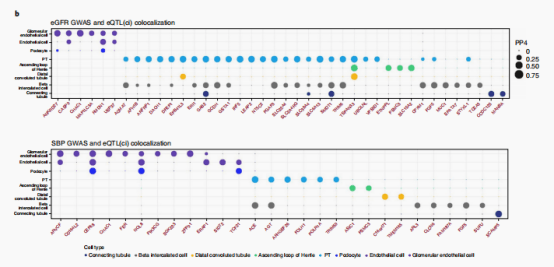
总的来说,作者基于全面的多组学数据集,并使用正交分析方法来注释与肾脏相关的表型。这些分析优先选择出了超过182个可能的肾功能致病基因和88个高血压致病基因,这些可以作为肾脏中这些疾病的核心基因。
从文章中可以看出,基于单细胞结合其他组学的分析,对于挖掘相关基因,精细筛选相关基因是十分有效的。
转自:派森诺
- 本文固定链接: https://maimengkong.com/zixun/1506.html
- 转载请注明: : 萌小白 2023年5月2日 于 卖萌控的博客 发表
- 百度已收录
