
许多流行病的爆发都是病毒引起的,面对新的传染性基因组出现的最佳策略是及时识别,以便于在感染开始时立即实施相应措施。
目前可用的诊断测试仅限于检测新的病理因子。适用于同时检测存在的任何病原体的高通量方法可能比使用基于当前方法的大量单独测试更有优势。
宏基因组学测序、全基因组测序和靶向深度测序是目前用于病毒遗传鉴定和表征的最佳工具。通过使用这些技术,可以正确的对病毒进行分类,确定其变异性,识别与毒性相关的病毒遗传标记,并在现有知识的基础上考虑抗原性和对抗病毒药物的易感性。
尽管宏基因组学领域取得了巨大进步,但对于具体数据分析任务应使用各种方法中的哪一种,仍缺乏共识。

本文重点描述了宏基因组生物信息数据处理所需要的工具,以便于改善使用宏基因组学识别动物来源样本中新出现、再出现和未知的新病毒。
宏基因组学识别病毒数据处理流程工具
什么是宏基因组学?
宏基因组学是下一代测序的一个领域,可以识别微生物群落,以及基因检测、识别和表征致病因子。它已被证明是病毒遗传特征的关键因素,并导致了使用传统培养技术无法完成的发现。
目前的分子检测使用特定的引物或探针针对有限数量的病原体,而宏基因组学可以接近样本中存在的所有 DNA 和 RNA 分子,从而能够分析相应的宿主基因组及其微生物集合。
在宏基因组组装中鉴定病毒有五个主要步骤:
- 质量控制(QC)
- 修剪低质量序列
- 组装
- 组装后的数据质检(可选)
- 对组装后序列进行物种注释(识别已经测序的已知病毒和识别尚未测序或未知的病毒)
宏基因组分箱是在物种注释之前可选的附加步骤。分箱的目的是根据根据序列的起源对其进行聚类。
根据这些步骤,列举出以下目前使用较多的主流工具。
序列质检工具
宏基因组学的第一步将是执行序列QC,因为从分析中消除技术错误是必不可少的。
此步骤的主要目的是识别不需要的接头序列、过短的序列、低质量的序列或核苷酸以及其他可能存在的数据。根据数据类型,在这一步中可以使用以下几种工具:

对于短读,可以使用FastQC执行质检 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) ,它可以检查序列的质量并生成总结报告。
其他QC程序也可以提供相同类型的报告,如MultiQC,它具有与FastQC相同的功能,但有一个主要区别,它可以同时合并多个fastq的QC报告,生成一个总的报告。
对于长读,可以使用longQC或MinionQC来检查序列质量,这两个工具已经应用于从纳米孔的MinION或其他长读取测序仪中获得的数据。
用于数据预处理的工具
—— 低质量序列修剪工具
序列质检后,就需要修剪工具,删除低质量序列和接头序列。可供使用的工具如下图。

对于短读,常用的是Trimmomatic,其次是Cutadapt和Fastp。
对于长读,NanoPack可用于处理长读数据并可视化QC结果。与Nanopack功能相同的是SequelTools。
——删除测序数据中非靶向或污染序列的工具
删除不感兴趣的序列,这些序列可以从各种来源获得。在对病毒序列的分析中,必须删除宿主序列和被污染序列,它可以减少假阳性,并可以防止嵌合病毒-宿主序列的组装。
如下图,通常使用序列比对的工具:

对于短读,可以使用BWA、bowtie2和BBMap等。
其它工具如FastQ-Screen,可以以fastq格式比对自定义参考序列。
(https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/)
对于长读,可以使用BWA和BBMap,也可以选择特定的minimap2。
也有专门用于识别和修剪特定微生物类群序列的工具,这类工具通常已包含参考基因组序列,一旦比对上,将通过内置的过滤程序丢弃掉。比如VirusHunter(https://bio.tools/virushunter),用于识别NGS数据中的病毒序列。
某些情况下,可能需要从宏基因组数据中删除非靶向分类群的其他RNA序列类型,如核糖体(rRNA),线粒体(mtRNA),或mRNA类型。这时可以用RiboDetector (https://github.com/hzi-bifo/RiboDetector),因为它专门识别rRNA,从而可以过滤掉rRNA以改进后续分析。
另一种方法是在组装前对序列进行物种注释。使用这种策略,可以过滤掉病毒以外的序列,保留病毒序列以供进一步分析。可以使用kraken2和kaiju。
序列组装
为了更好的进行物种注释和识别存在的病毒,对序列进行组装,生成contigs,以提供更长的连续序列。宏基因组学中使用的组装类型主要为de novo,即从头基因组组装。
可使用的工具如下图:
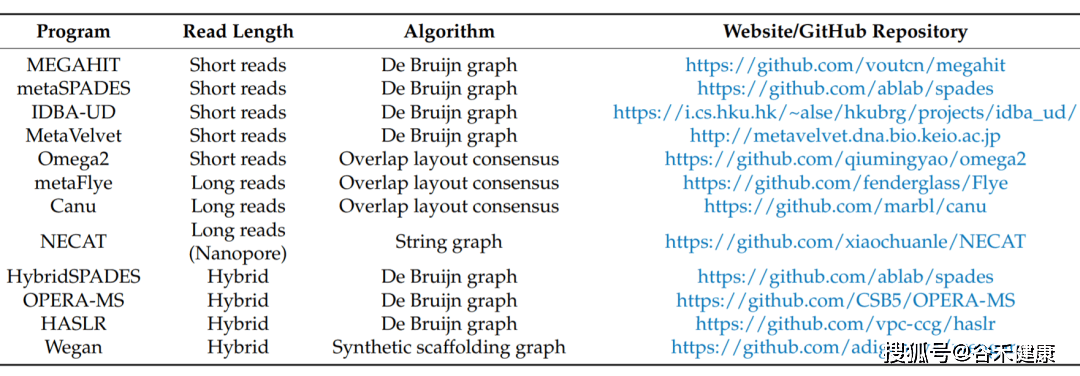
对于短读,推荐MEGAHIT,这是一个针对宏基因组优化的生物信息学组装工具,或者metaSPADES和IDBA-UD,它们也针对宏基因组进行了优化。
除了de novo,还有一种基于参考的组装,也可以用于宏基因组学。只是,并不是在所有情况下都可以获得合适的参考基因组,而且这种方法不能识别新的病毒或以前没有测序的病毒。
对于长读,推荐metaFlye、Canu和NECAT,这些工具可以用于各种技术下产生的数据格式,从纳米孔测序到PacBio,甚至在高保真序列。
对于混合组装,也就是将短读和长读的两个特性结合起来的组装,推荐OPERA-MS和HybridSPADES工具,它们都是用De Bruijn图算法实现的。
对组装完成后的序列质检
宏基因组组装完成,就应该确定组装的质量。用于此目的的工具可以分为两大类:
一类是需要参考基因组的工具,例如MetaQUAST,它使用参考来计算组装的统计信息。一般而言,在宏基因组学研究中,可能很难使用参考基因组,因为通常没有可用的参考基因组或参考基因组的质量很差。
不需要参考基因组的方法,例如DeepMAsED,它使用机器学习来识别错误装配,或者REAPR,是一种使用映射的配对端读长来评估基因组组装准确性的工具。常用的还有BUSCO和CheckM。
最后,VALET(https://github.com/marbl/VALET)可以用于检测宏基因组数据中的误组装,因为它可以根据覆盖范围对contigs进行分类,并避免由于覆盖深度不均匀而导致的假阳性和假阴性。

在宏基因组分析中识别病毒的一个重要步骤是进行物种注释。实现这一步骤的主要方法有两种:
- 第一种是直接对序列进行分类
- 第二种是通过contigs建立分类
两种方法各有优缺点:
在使用contig(即使用组装序列)进行的物种注释中,分类的对象是较长的序列,它存在一些contigs可能是嵌合的风险。
而直接对reads进行物种注释的统计学意义较小,虽然分析了大量的序列,但序列较短,这种方法可以提供更多样化的结果,只是计算成本会更高。
识别已知病毒或未知的新病毒的工具
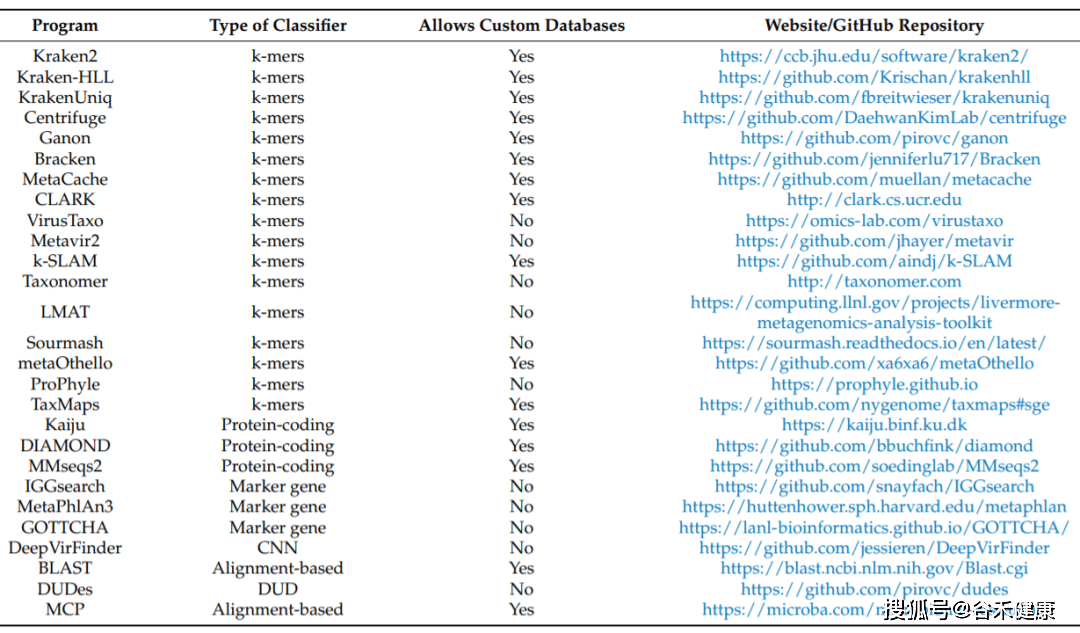
对于已知病毒的识别,一种是基于k-mer,直接使用参考数据库与reads/contigs进行比对,如kraken2、bracken、CLARK和Centrifuge,
另一种是先翻译序列,然后与参考蛋白质数据库进行比对,如kaiju、DIAMOND和MMseqs2。
还有基于算法的,如BLAST或DUDes,它们使用DUD(Deepest Uncommon Descent)算法。
使用基因标记的,如MetaPhlAn4、IGGsearch和GOTTCHA。
也有专门用来研究病毒组的工具,如VirusTaxo、Metavir2和DeepVirFinder,其主要算法是卷积神经网络(CNN)。
其中如MetaPhlAn4和MCP (Microbiota Community Profiler),包含未知的宏基因组组装基因组的序列,而MCP只能用于识别微生物区研究中的细菌、古菌、真核生物和病毒序列。
由于每个用于物种注释的工具的性能都不同,且都使用了各种算法和参考数据库,所以这种多样性也会导致不一样的结果、耗时和计算成本。
▪ 基于k-mer的物种注释工具似乎是计算效率最高的,虽然它们需要很大的内存。
▪ 基于标记的,对内存的要求较低,但它们只能对来自特定区域的reads/contigs进行注释分类。
▪ 基于比对的工具要比其他的计算成本更高。
对于新型病毒的识别,现在也有不需要任何参考就能识别病毒序列的工具,即:
• VirSorter(https://github.com/simroux/VirSorter)
• VirFinder(https://github.com/jessieren/VirFinder)
VirFinder是一个基于k-mer的R包,可以以较好的预测识别病毒的contigs;
而VirSorter可以在不同的微生物数据集中识别新的病毒序列。
宏基因组分箱
在物种注释之前可以选择是否执行分箱(binning)。
分箱的主要目的是根据物种对contig进行聚类。根据数据类型,可使用的工具如下图:

CONCOT,它可以根据核苷酸组成和覆盖率数据对宏基因组contigs进行聚类。
GraphBin,它使用组装的连通性信息对contig进行集群化。
但宏基因组分箱并不局限于contigs,对于长读,可使用MEGAN-LR、BusyBee或LRBinner。
结 语
近年来,宏基因组学领域取得了许多进展,新技术可以帮助研究人员发现新的病毒,预测疫情,诊断某些疾病等。
长读测序平台也在快速发展,以得出更可靠的结果助力宏基因组分析。虽然已有许多工具和流程被开发出来以便更快更简单地进行数据分析,但还需要进一步发展,例如在数据处理分析中的通用指南的建立,因为虽然出于同一种目的而开发的工具,但由于计算过程不一样,它们在不同任务中的性能缺乏共识。此外,重要的是保持相关数据库的更新与维护。
宏基因组学检测人类样本中任何基因组(包括细菌、病毒、寄生虫和真菌)的能力,对于传染病的诊断具有重要意义。宏基因组学方法也已应用于其他几个研究领域:环境研究(如海洋样本、土壤、污水、农场灰尘) ;7000 年前青铜时代人类样本中的病毒感染;健康、疾病和法医调查中人体肠道微生物组的特征;临床研究 ; 以及新病毒病原体的发现,例如 SARS-CoV-2等。
参考文献:
Ibañez-Lligoña M, Colomer-Castell S, González-Sánchez A, Gregori J, Campos C, Garcia-Cehic D, Andrés C, Piñana M, Pumarola T, Rodríguez-Frias F, Antón A, Quer J. Bioinformatic Tools for NGS-Based Metagenomics to Improve the Clinical Diagnosis of Emerging, Re-Emerging and New Viruses. Viruses. 2023 Feb 20;15(2):587. doi: 10.3390/v15020587. PMID: 36851800; PMCID: PMC9965957.
- 本文固定链接: https://maimengkong.com/kyjc/1489.html
- 转载请注明: : 萌小白 2023年4月28日 于 卖萌控的博客 发表
- 百度已收录
