声明:本文转载自“微生物生态”公众号,一个有干货的公众号
系统发育树
系统发育树(Phylogenetic tree)又称为系统进化树,是用一种类似树状分支的图形来概括各物种之间的亲缘关系,可用来描述物种之间的进化关系。
1.系统发育树构建步骤
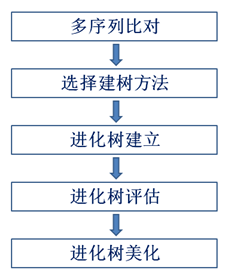
2.多序列比对
系统发育树构建的第一步是进行多序列比对,常用的软件包括MEGA, cluster X,Muscle,phylip等。(都很常用,就看哪个顺手)
MEGA是最常用的比对建树软件,优点是可视化图形界面,简单方便;缺点是比对速度慢,输出格式单一。
Cluster X 的优点是图形界面,可输出多种格式(如phy);缺点也是慢。
Muscle和phylip 的优点是运算快,不过需要输入简单地代码,可能不适合初学者。
3.选择建树方法
系统发育树构建的基本方法有如下几种:
1、Distance-based methods 距离法:
(基于距离的方法:首先通过各个物种之间的比较,根据一定的假设(进化距离模型)推导得出分类群之间的进化距离,构建一个进化距离矩阵。进化树的构建则是基于这个矩阵中的进化距离关系。)
· Unweightedpair group method using arithmetic average(UPGMA)非加权分组平均法
· Minimum evolution(ME)最小进化法
· Neighbor joining(NJ)邻位归并法
2、Character-based methods 特征法:
(基于特征的方法:不计算序列间的距离,而是将序列中有差异的位点作为单独的特征,并根据这些特征来建树。)
· Maximum parsimony(MP) 最大简约法
· Maximum likelihood method(ML) 最大似然法
模型选择的依据如下图:
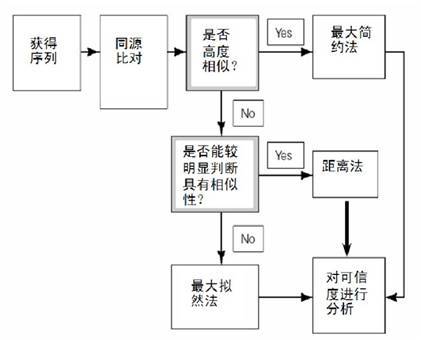
其中UPGMA法已经较少使用。一般来讲,如果模型合适,ML的效果较好。对近缘序列,有人喜欢MP,因为用的假设最少。MP一般不用在远缘序列上,这时一般用NJ或ML.对相似度很低的序列,NJ往往出现Long-branch attraction(LBA,长枝吸引现象),有时严重干扰进化树的构建。贝叶斯的方法则太慢。对于各种方法构建分子进化树的准确性,一篇综述(Hall BG. Mol Biol Evol 2005,22(3):792-802)认为贝叶斯的方法最好,其次是ML,然后是MP。其实如果序列的相似性较高,各种方法都会得到不错的结果,模型间的差别也不大。不过现在文章普遍使用的是NJ是ML模型。
4.进化树评估
用截然不同的距离矩阵法与简约法分析一个数据集,如果能产生相似的系统发育树,这样的树可以认为是可靠的。
我们一般用Bootstrap(自展法)进行检验,现在一般文章要求Bootstrap值1000。虽然根据严格的统计学概念,自展值要大于95%才较为可信,然而在实际应用中,特别是微生物等相似度比较大的分类中,一般大于50%就认为可信(小于50%隐去)。
系统发育树的一般解读如下图:
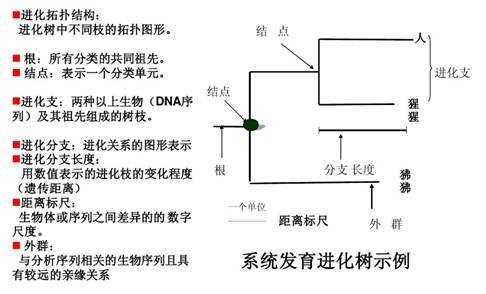
对我们而言 ,最常用到的三个指标分别是距离标尺,分支长度和自展值。
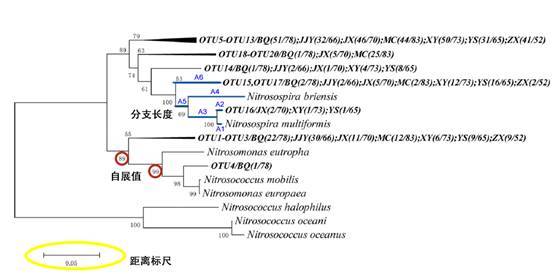
距离标尺:进化树可以显示序列的差异度,这里的标尺就可以当做为进化树的“比例尺”。
分支长度:在树形结构中,枝长累积距离越近的样本差异越小,反之差异越大。比如OTU16与Nitrosospira multiformis的差异度是A1+A2,OTU16与Nitrosospira briensis的距离是A2+A3+A4,以此类推。
自展值:刚才已经讲过关于自展值的评估方法。自展值可以显示可信度。一般低于50%的会隐去。那啥情况下会低于50%呢,两种情况,相似度太低或太高。一般来说,低自展值靠近分支末端,可能是由于相似度太高难以区分,这时建议可以换一个基因建树。如果低自展值靠近根,可能是由于相似度太低。
5.进化树美化
进化树没问题以后,可以在美学角度对进化树进行改善。可以用到的软件有AI, PS,ggtree、GraPhlAn、treeview,Figtree,和在线网站ITOL等。一般需要建完树后用输出格式为 *.tree 或 *.nwk 的文件,导入到相关软件,进行修饰。
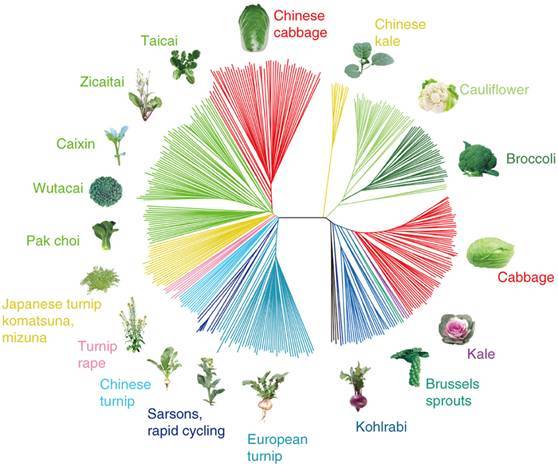
最后,就可以做成这样的图啦!

- 本文固定链接: https://maimengkong.com/kyjc/1458.html
- 转载请注明: : 萌小白 2023年4月17日 于 卖萌控的博客 发表
- 百度已收录
