很多小伙伴拿到转录组结果,都会有一个大大的疑问,明明上一步已经计算了FPKM了,为什么差异分析还是用readcount来做,我想用FPKM行不行?
这里,郑重地回答你:不可以且没必要!你熟悉的DESeq,DESeq2和EdgeR都表示不同意!他们都只认readcount!
首先,先让你和老朋友FPKM再重新熟悉一下:由于不同样品过滤后获得的数据量是不可能完全一致的,不同基因长度也有很大差异。因此为了能够在样品内比较基因的表达量,需要采用FPKM 对表达量进行标准化(Normalization):FPKM(Fragments Per Kilobase Million),为每百万 Reads 中来自某一基因每千碱基长度的 Reads 数目,是一种普遍采用的基因表达量标准化方法,这种方法同时考虑了测序深度和基因长度对基因表达量计数的影响。其 计算公式 如下:
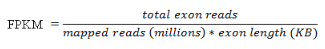
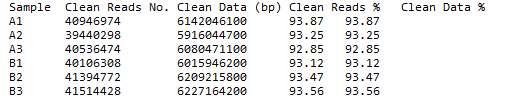
敲黑板!FPKM考虑到基因长度对基因表达量计数的影响,但是在进行差异分析时,同一个基因在不同样本中的表达差异根本不需要考虑这条基因的长度!!!差异分析需要均一化的是不同处理中的基因,FPKM均一化的则是这个样本中的所有基因,硬要让FPKM去做不属于他的工作,小心出现大问题!口说无凭,这里我们随机抽取了同一个项目的两组幸运数据,每组3个重复,直接来验证一下:
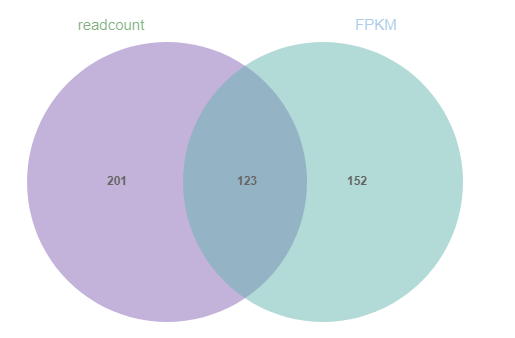
同样的两组数据,我们分别用readcount和FPKM进行差异分析,分别得到了324个和275个“差异基因”,光看数目是不是好像差别不大?可是这些基因中,只有123个是相同的,所以如果你用FPKM强行做了差异分析,那么得出的结果,可与用readcount的差异分析结果大相径庭。你以为得到的是孙悟空,其实根本就是披着孙悟空马甲的六耳猕猴!
这里可能小伙伴又疑惑了起来,FPKM又不能做差异,我要他有什么用???
No No No,想要比较同一个样本中所有基因谁的表达量更高更强,还是要FPKM出马。以及你熟悉的样品相关性分析、热图和WGCNA,他们通通都需要FPKM的支持!
因此,分析中无论是选用FPKM,还是readcount,都是经过统计学家,软件开发人员和分析人员的反复验证,深思熟虑选用的,所以如果你的结果还是不太满意,是不是也想想实验?
以上结果仅供参考!- 本文固定链接: https://maimengkong.com/kyjc/1242.html
- 转载请注明: : 萌小白 2022年10月5日 于 卖萌控的博客 发表
- 百度已收录
