本期,我们讲讲甲基化RNA免疫共沉淀(MeRIP-seq/m6A-seq)实验怎么做,从技术原理、建库测序流程、信息分析流程和研究套路等四方面详细介绍。
一、甲基化RNA免疫共沉淀(MeRIP-seq/m6A-seq)测序技术原理
表观转录组指RNA序列不发生改变的情况下,由RNA上的化学修饰调节基因表达的现象。胞内RNA的修饰超过100种,其中大部分的表观修饰发生在tRNA及其他非编码RNA上[1],发生在mRNA上的修饰无论是从种类上还是数量上都较少。下图展示了部分发生在真核生物mRNA上的化学修饰[2]:
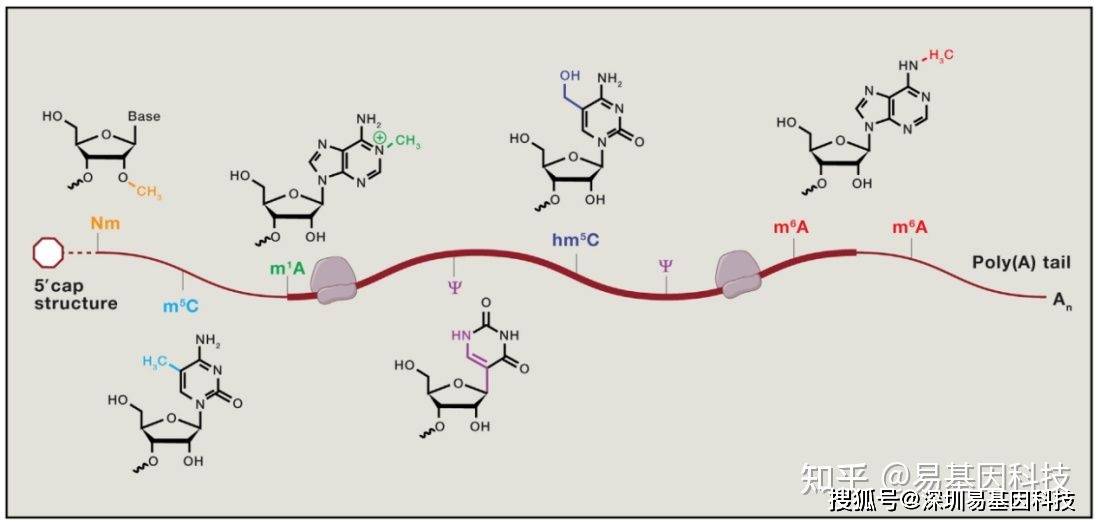 图1: mRNA甲基化修饰示意图
图1: mRNA甲基化修饰示意图
mRNA上含量最高的修饰是腺苷酸N6位的甲基化修饰(N6-methyladenosine,m6A),m6A参与mRNA生命周期的各个方面[2]。m6A修饰主要发生在RRACH (R = G or A and H = A,C, or U)这类motif上,这类motif主要在终止密码子和3’UTR处富集,即m6A主要发生在终止密码子和3’UTR附近。m6A修饰由m6A甲基转移酶(writers)催化产生,被m6A去甲基酶(erasers)去除,被m6A结合蛋白(readers)识别并发挥功能[3]。m6A修饰广泛存在于各类生物中,包括病毒、从酵母、植物和人类等高等动物[1,3]。
研究表明,m6A已知的最主要功能是调控mRNA的稳定性:胞质内经过m6A修饰的mRNA可以被YTHDF2识别,使其富集到Processing body(P-body)从而加速mRNA的降解。此外,m6A修饰还可以改变RNA的二级结构、调控microRNA的靶标识别来调控mRNA的稳定性。在核内,m6A修饰可以调控RNA的剪接、出核过程,从而调控基因表达。m6A还可能与DNA甲基化存在互作关系。下图展示了部分目前已知的m6A与生物功能的关系:
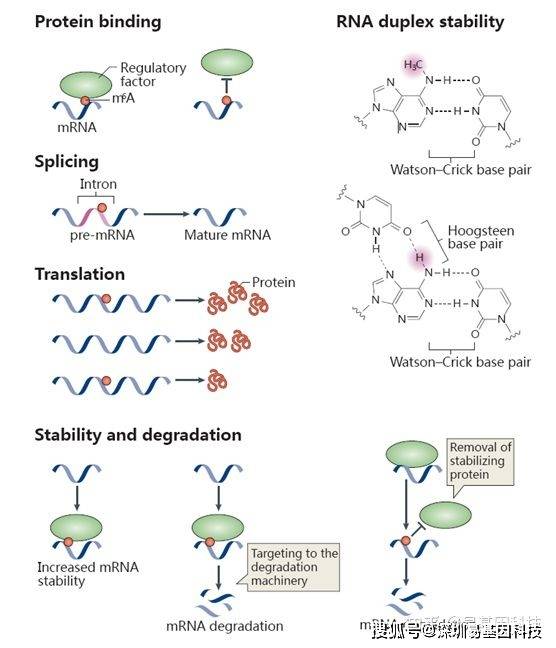 图2:m6A与生物学功能
图2:m6A与生物学功能
早期的实验方法已经在rRNA和mRNA中鉴定到m6A的存在,但由于实验技术的限制,对m6A的研究一直没有大的进展。近年来,RNA去甲基化酶FTO的报道,使得对m6A修饰的研究再次进入人们的视野。随后,MeRIP-seq/m6A-seq实验方法的建立使得有机会对该修饰的分布和功能进行深入研究[1]。
利用MeRIP-seq/m6A-seq技术鉴定全基因组范围内具有m6A修饰的区域。其原理为通过特异识别m6A修饰的抗体,对细胞内具有m6A修饰的RNA片段进行免疫共沉淀。对沉淀下来的RNA片段进行高通量测序,结合生物信息学分析,即可在全基因组范围内对m6A修饰的状况进行系统研究。MeRIP-seq/m6A- seq是目前研究m6A修饰使用最广泛的技术之一。
二、甲基化RNA免疫共沉淀(MeRIP-seq/m6A-seq)实验流程
从样品处理到最终数据获得中每一个环节都会对数据质量和数量产生影响,而数据质量又会直接影响后续信息分析的结果。为了从源头上保证测序数据的准确性、可靠性,需对样品处理、建库、测序每一个生成步骤都严格把控,从根本上保证了高质量数据的产出。流程图如下:
(一)Total RNA样品检测
对RNA样品的检测主要包括3种方法:
(1)琼脂糖凝胶电泳分析RNA降解程度以及是否有污染,检测具有明显的18S或28S主带,且条带清晰;
(2)Qubit 2.0对RNA浓度进行精确定量,总RNA 检测总量不低于10ug;
(3)Agilent 2100精确检测RNA的完整性,RIN值不低于7.5;
(二)文库构建
1. RNA片段化:
取10ug总RNA,加入RNA Fragmentation Reagents(Invitrogen)在Thermomixer中70℃反应10min,将RNA打断成片段,片段大小约为100nt,用乙醇法将片段化的RNA沉淀;
2. m6A富集:
将含protein A和protein G的磁珠用IP buffer(150 mM NaCl, 10 mM Tris-HCl pH 7.5)洗涤,然后与5 ug m6A抗体(Millipore) 4℃孵育2h;用 IP buffer洗涤两次,再用 IP buffer将磁珠重悬,加入片段化的RNA,4℃下翻转4h;用IP buffer在4℃下洗涤磁珠3次,再用m6A竞争性洗脱液,在4℃下孵育1h。将含有洗脱m6A RNA的上清收集到新的试管中,用苯酚:氯仿:异戊醇(125:24:1)进行纯化;
3. 文库构建:
用SMARTer® Stranded Total RNA-seq Kit v2 - Pico Input Mammalian User Manual根据说明书对IP与Input样品进行反转录及文库构建;利用AMPure XP bead进行片段大小选择,获得最终文库。
构建原理图如下:
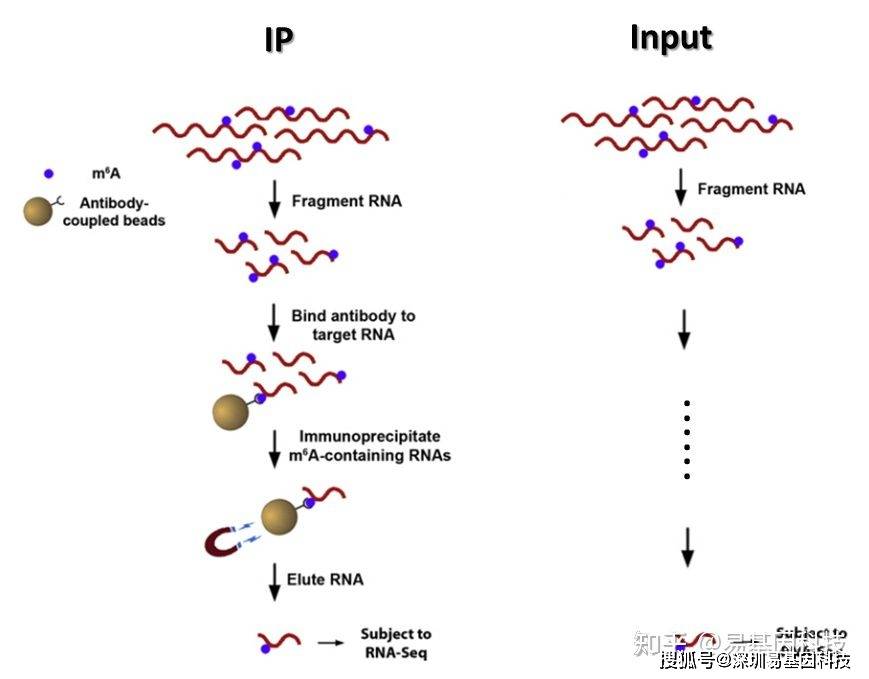 图3:富集流程示意图
图3:富集流程示意图
(三)文库质检
文库构建完成后,先使用Qubit2.0进行初步定量,稀释文库至1ng/ul,随后使用Agilent 2100对文库的insert size进行检测,insert size符合预期后,使用qPCR方法对文库的有效浓度进行准确定量(文库有效浓度> 2nM),以保证文库质量。
(四)上机测序
文库检测合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后在illumina Nova平台测序,测序策略为PE150。
三、甲基化RNA免疫共沉淀(MeRIP-seq/m6A-seq)信息分析流程
(一)原始下机数据质控
原始下机数据为FASTQ格式,是高通量测序的标准格式。FASTQ文件每四行为一个单位,包含一条测序序列(read)的信息。该单位第一行为read的ID,一般以@符号开头;第二行为测序的序列,也就是reads的序列;第三行一般是一个+号,或者与第一行的信息相同;第四行是碱基质量值,是对第二行序列的碱基的准确性的描述,一个碱基会对应一个碱基质量值,所以这一行和第二行的长度相同。以下为一条read信息的示例:
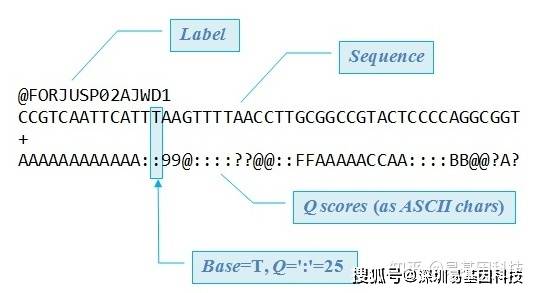 图4:FASTQ格式示例
图4:FASTQ格式示例
原始下机数据包含建库时引进的接头序列以及质量过低的碱基,这些因素会导致后续比对到基因组的reads较少,从而导致得到的信息较少,因此需要进行过滤。
利用Trimmomatic软件对原始数据进行去除接头序列及低质量碱基等质控步骤,所用参数为:ILLUMINACLIP:MeRIP-PE.fa:2:30:10:1:true SLIDINGWINDOW:30:15 AVGQUAL:15 LEADING:15 TRAILING:15 MINLEN:30
(二)数据比对
过滤后的数据需要比对至参考基因组,基因组上发生m6A修饰的区段会有大量reads比对上去,从而形成“峰”(peak),根据峰的位置即可判断基因组的哪些位置发生了甲基化。
比对采用hisat2[4],该软件可快速将短序列比对至参考基因组,并且可以考虑并处理拼接位点,尤其适用于RNA数据的比对。所用参数为默认参数。比对完成后,对结果路径进行去除多重比对、去除低质量比对等过滤,从而得到更精确的比对结果。
(三)检测m6A修饰区域
MeRIP-seq将RNA上发生m6A修饰的区域富集后进行测序,因此发生m6A修饰的区域,IP文库所覆盖的reads数会显著高于input文库,从而形成“峰(peak)”。检测这些峰的位置即可得到RNA上发生m6A修饰的位置。
鉴定得到m6A修饰后,再对m6A修饰进行注释、分布统计、motif鉴定等分析。
(四) 差异m6A修饰区域的鉴定
差异m6A修饰(即差异peak)的鉴定采用R包exomePeak[5,6]。该R包先将需要比较的样本的peak进行合并,再计算每个样本的合并后peak内累积的reads数目;再对这些reads进行标准化,使两组样本处于可比较的水平;最后检验两组样本在该peak内的reads数目是否有显著差异。所用参数为:WINDOW_WIDTH=200 SLIDING_STEP=30 DIFF_PEAK_ABS_FOLD_CHANGE = 1.5 FOLD_ENRICHMENT=1.5 FRAGMENT_LENGTH=200。
鉴定差异m6A修饰后,再对差异m6A修饰进行注释、分布统计、motif鉴定等分析。
(五)mRNA基因表达水平分析
m6A-seq的input文库相当于RNA-seq文库,可以用于分析基因表达量及鉴定差异表达基因。通过定位到基因组区域或者基因外显子区的测序序列(read)的计数来估计基因的表达水平。Read计数除了与基因的真实表达水平成正比外,还与基因的长度和测序深度成正相关。为了使不同基因、不同实验间计算的基因表达水平具有可比性,利用TPM来对read数量进行标准化,该算法考虑了测序深度和基因长度对计数的影响,是目前最为常见的基因表达水平计算方法之一。
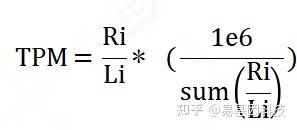
图5:TPM计算公式
注:Ri表示该基因的read count数目;Li表示该基因的长度(kbp),选取最长的转录本作为该基因的长度。表达量计算软件为StringTie[7],使用默认参数,用TPM展示基因表达量。
(六)lncRNA筛选鉴定及表达量计算
通过对转录本进行组装,可以发现注释文件外的新转录本。对这些新转录本进行一系列严格条件筛选,鉴定出非编码RNA。
(1)使用软件StringTie[7]对转录本进行组装,获得每个样品中所有的转录本,将没有在注释文件中出现的转录本标记为新转录本。对新转录本筛选条件为:a)对于每个样品的组装注释文件,过滤掉转录本exon个数 >=2 但FPKM<=0.5或者exon个数>=1但FPKM<=1的转录本[8];b)过滤单个转录本拼接结果中长度低于200bp的转录本;c)将以上单个过滤后的组装转录本用StringTie[7]合并成最终组装转录本;d)通过Cuffcompare[9]软件分析未知转录本与已知转录本的结构关系。
(2)候选lncRNA编码能力预测。lncRNA是一类非编码RNA,通过预测候选lncRNA的编码能力,进一步鉴定出真正的lncRNA。采用多款主流编码能力预测软件对候选lncRNA进行编码能力预测。预测软件包括CNCI[10]和CPC[11]。
(3)针对基因组有注释的lncRNA基因信息,计算known lncRNA的表达情况。
(4)表达量计算。用TPM标准化lncRNA的表达量。
(七)circRNA鉴定及表达量计算
采用两款主流软件进行circRNA鉴定[12],最后取两款软件并集作为最终预测结果。该两款软件分别为find_circ[13]和CIRCexplorer2[14]。
(1)Find_circ预测原理如下图所示:
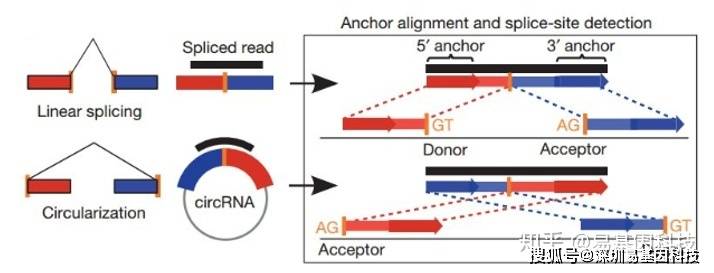 图6:find_circ软件鉴定原理
图6:find_circ软件鉴定原理
基本原理是从没有比对到参考序列的 reads 的两端各提取 20-nt 的 anchor 序列,将每一对 anchor 序列再次比对参考序列,如果 anchor 序列的 5' 端比对到参考序列(起始与终止位点分别记为 A3,A4),同时该 anchor 序列的 3' 端比对到此位点的上游(起始与终止位点分别记为 A1,A2),并且在参考序列的 A2 到 A3 之间存在剪接位点(GT-AG),则将此 read 作为候选 circRNA。最后将 read count 大于等于 2 的候选 circRNA 作为鉴定的 circRNA。比对所采用软件为bowtie2[15]。
(2)CIRCexplorer2测原理如下图所示:
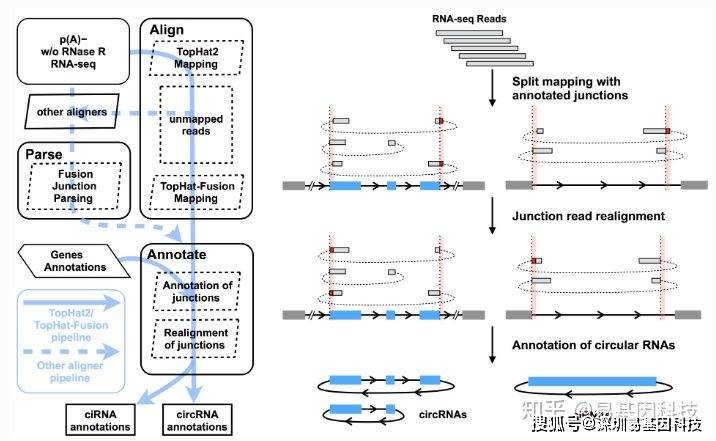 图7:CIRCexplorer2软件鉴定原理
图7:CIRCexplorer2软件鉴定原理
该软件可支持TopHat2/TopHat-Fusion,STAR,MapSplice,BWA和segemehl等多种RNA aligners。采用STAR[16]软件进行比对。
表达量计算:由于计算的是circRNA anchor 序列的read count值,因此,进行标准化时候采用SRPBM[17]对circRNA进行表达量计算。公式为:
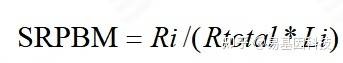
图8: SRPBM 表达量计算公式
注:Ri表示该circRNA anchor 序列的read count数目;Li表示该circRNA anchor 序列的长度(k bp)。
(八)差异表达基因鉴定
在获得各样品的基因表达量后,为了确定处理组相比对照组有哪些基因表达量发生了较大变化,对各组样品进行差异基因分析。根据各组样品的基因的read count,可采用目前使用最广的差异基因分析软件进行差异基因分析,有生物学重复分析软件为DESeq2[18],无生物学重复分析软件为edger。
(九)差异表达基因富集分析
利用clusterprofiler对差异表达mRNA、差异表达cicrRNA的源基因、差异表达lncRNA的靶基因分别进行GO和KEGG富集分析并作图。
(十)差异m6A关联基因与差异表达基因重叠分析
为了了解差异m6A关联基因与差异表达基因之间的重叠关系,进行如下统计:
(1)得到每个基因关联的差异peak(包括显著和不显著的),一个基因可能对应多个peak,这种情况下这个基因将出现多次;也可能没有对应的差异peak,这种情况下该基因将不会出现。
(2)鉴定(1)中得到的基因与差异表达基因之间的重叠关系。
(3)利用四象限图展示。
(十一)信息分析流程示意图
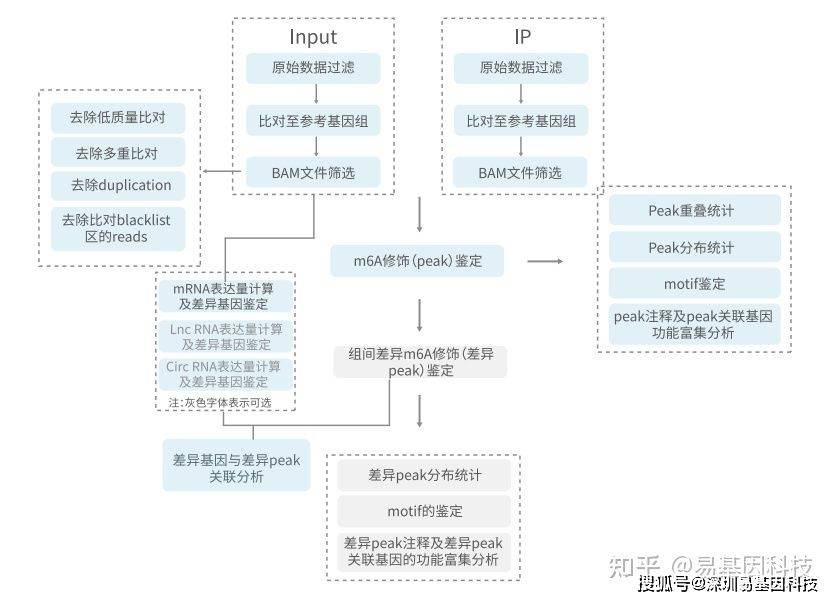 图9:分析流程示意图
图9:分析流程示意图
四、总结:甲基化RNA免疫共沉淀(MeRIP-seq/m6A-seq)研究思路
MeRIP通过m6A特异性抗体富集和测序,用于研究RNA的腺苷甲基化修饰。易基因自主研发微量RNA甲基化检测技术,样本起始量可降低至10-20μg,最低仅需5μg总RNA。
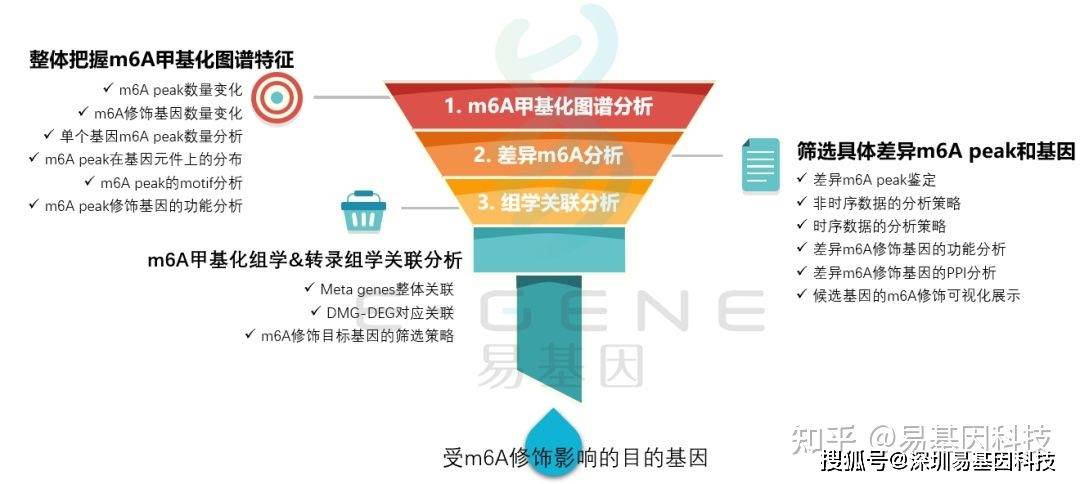
m6A研究思路
- 整体把握m6A甲基化图谱特征:m6A peak数量变化、m6A修饰基因数量变化、单个基因m6A peak数量分析、m6A peak在基因元件上的分布、m6A peak的motif分析、m6A peak修饰基因的功能分析;筛选具体差异m6A peak和基因:差异m6A peak鉴定、非时序数据的分析策略、时序数据的分析策略、差异m6A修饰基因的功能分析、差异m6A修饰基因的PPI分析、候选基因的m6A修饰可视化展示;m6A甲基化组学&转录组学关联分析:Meta genes整体关联、DMG-DEG对应关联、m6A修饰目标基因的筛选策略;进一步验证或后期试验。
五、甲基化RNA免疫共沉淀(MeRIP-seq/m6A-seq)研究项目案例

标题:Sevoflurane impairs m6A-mediated mRNA translation and leads to fine motor and cognitive deficits七氟烷麻醉破坏m6A介导的mRNA翻译并导致精细运动和认知障碍
时间:2021
期刊:Cell Biology and Toxicology
影响因子:IF 6.691
技术平台:m6A-seq(MeRIP-seq)、RIP-seq (RNA结合蛋白免疫沉淀) 、scRNA-seq
研究摘要:
本研究探索了全麻药物对婴幼儿精细运动能力损伤的机制,并首次关注了m6A甲基化在七氟烷麻醉影响精细运动损伤中的作用和机制。
以前的研究发现全麻药物引起的神经发育毒性机制在非人灵长类和啮齿类动物模型之间尚存在一定的差异,比如临床上观察到多次全麻和手术引起婴幼儿患者远期语言和社交能力的降低和非人灵长类动物模型观察到麻醉后多种社交行为能力的损害,但是多次的麻醉的幼鼠却很难观察到社交能力的损害。研究人员认为在全麻药物引起认知损伤的非人灵长类模型和啮齿类模型中,肯定有同向变化的基因,也肯定有相反变化的基因,应该在临床上找寻出引起全麻药物神经发育毒性的关键科学问题,然后使用非人灵长类动物猕猴去探索全麻药物引起神经发育毒性的机制线索,之后找到猕猴和小鼠具有相同变化的靶基因,使用小鼠模型来进行验证。
基于这个理念,研究人员聚焦到了使用非人灵长类动物和啮齿类动物来研究全麻药物与术后远期精细运动损伤的机制。m6A修饰是在RNA甲基化修饰中最丰富的一种,YTHDF1是m6A甲基化的识别蛋白之一。最近的研究发现其可以参与神经认知的形成和发展。在随后的机制研究中,研究发现七氟烷麻醉后的幼年灵长类和啮齿类动物的大脑中m6A结合蛋白YTHDF1表达显著下调。单细胞转录组测序(scRNA-seq)发现sp8阳性的神经元中YTHDF1的表达下降在中间神经元中最为明显,而这部分神经元,后续会发育成VIP中间神经元。YTHDF1的功能主要是识别RNA上的甲基化位点。通过RIP实验(RNA结合蛋白免疫沉淀)和m6A-seq测序分析发现m6A被高度富集在突触素(Synaptophysin)的mRNA上,同时Synaptophysin的mRNA上有YTHDF1的结合位点。早先的研究发现Synaptophysin和全麻药物的神经发育毒性紧密相关。过表达YTHDF1可以回救七氟烷引起的幼年小鼠精细运动能力和认知功能障碍以及Synaptophysin的变化。研究认为YTHDF1是以m6A甲基化依赖性方式调控其下游靶基因Synaptophysin的表达,继而损伤小鼠的精细运动能力和认知功能。本研究探索了全麻药物对婴幼儿精细运动能力损伤的机制,有望为预防或治疗全麻药的神经发育毒性提供新的思路。
 图1:研究摘要
图1:研究摘要
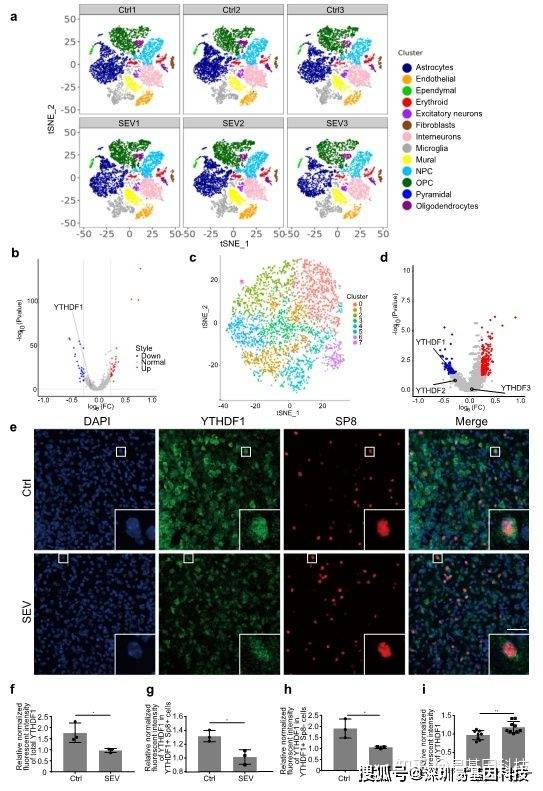 图2:scRNA-seq揭示七氟烷降低了中间神经元中YTHDF1的表达
图2:scRNA-seq揭示七氟烷降低了中间神经元中YTHDF1的表达
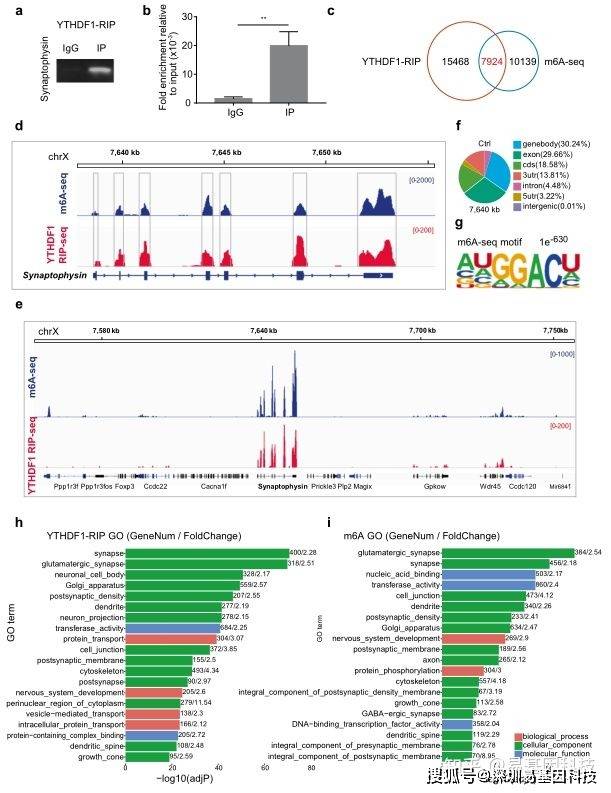 图3:MeRIP-seq揭示YTHDF1以m6A依赖的方式调节突触素
图3:MeRIP-seq揭示YTHDF1以m6A依赖的方式调节突触素
以上就是关于甲基化RNA免疫共沉淀(MeRIP-seq/m6A-seq)实验流程和分析思路的介绍,易基因科技提供全面的RNA甲基化研究整体解决方案。
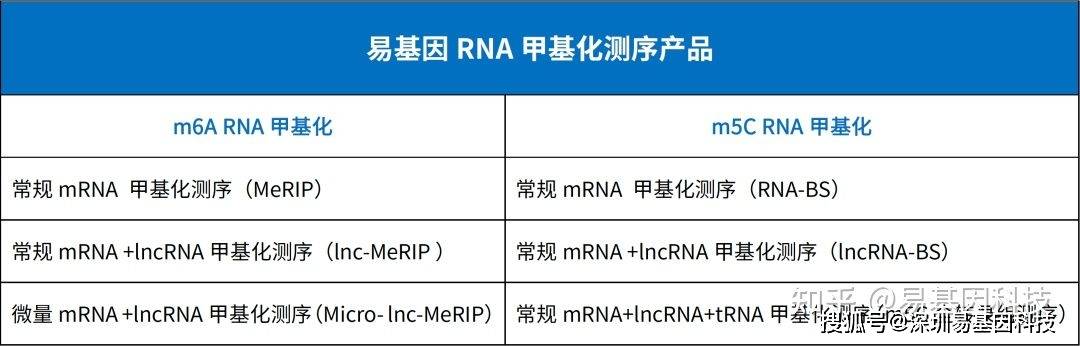
参考文献:
[1] Saletore Y, Meyer K, et al., The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biol. 2012 Oct 31;13(10):175.
[2] Roundtree IA, Evans ME, et al., Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017 Jun 15;169(7):1187-1200.
[3] Ma S, Chen C, et al., The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol. 2019 Nov 22;12(1):121.
[4] Kim D, Langmead B, et al., HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015 Apr;12(4):357-60.
[5] Meng J, Cui X,et al., Exome-based analysis for RNA epigenome sequencing data. Bioinformatics. 2013 Jun 15;29(12):1565-7.
[6] Meng J, Lu Z,et al.,A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package. Methods. 2014 Oct 1;69(3):274-81.
[7] Pertea M, Kim D, et al., Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016 Sep;11(9):1650-67.
[8] Yang, Y., et al., Recurrently deregulated lncRNAs in hepatocellular carcinoma. Nat Commun, 2017. 8: p. 14421
[9]Trapnell, C., et al., Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. 2010. 28(5): p. 511.
[10]Sun, L., et al., Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res, 2013. 41(17): p. e166.
[11]Kong, L., et al., CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res, 2007. 35(Web Server issue): p. W345-9.
[12]Hansen, T.B., et al., Comparison of circular RNA prediction tools. Nucleic Acids Res, 2016. 44(6): p. e58.
[13]Memczak, S., et al., Circular RNAs are a large class of animal RNAs with regulatory potency. Nature, 2013. 495(7441): p. 333-8.
[14]Zhang, X.O., et al., Complementary sequence-mediated exon circularization. Cell, 2014. 159(1): p. 134-147.
[15]Langmead, B. and S.L. Salzberg, Fast gapped-read alignment with Bowtie 2. Nat Methods, 2012. 9(4): p. 357-9.
[16]Dobin, A., et al., STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 2013. 29(1): p. 15-21.
[17]Zheng Q,Bao C,et al,Circular RNA profiling reveals an abundant circHIPK3 that regulated cell growth by sponging mutiple miRNAs. Nat Commun. 2016 Apr 6;7:11215
[18] Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
[19] Zhang L, et al. Sevoflurane impairs m6A-mediated mRNA translation and leads to fine motor and cognitive deficits. Cell Biol Toxicol. 2022 Apr;38(2):347-369.
转自:易基因- 本文固定链接: https://maimengkong.com/kyjc/1243.html
- 转载请注明: : 萌小白 2022年10月5日 于 卖萌控的博客 发表
- 百度已收录
