一文学会利用ComplexHeatmap包绘制超美热图
大家好,我是风。随着生物信息学的普及,各类在线网站已经能够做出很多精美的图片。一种东西大众化,那想做得脱颖而出,就要考虑如何把大众化的东西做得个性化。尽管绘图网站能够做出精美的图片,然而却无法让你与众不同。好马要靠鞍,好数据也要有个好包装,今天开始,我们来一起学习一个绘制热图的R包——ComplexHeatmap包,用它来为你的文章添加一张让人耳目一新的美图。
图片预览
我们先来看看ComplexHeatmap绘制的热图:
好看吧?绘制热图的R包有很多,包括ggplot2、gplot、pheatmap等等,但是如果说哪个R包最全面并且绘制的图片层次最丰富,我会推荐你使用ComplexHeatmap包。ComplexHeatmap包是由顾祖光博士基于grid绘图系统开发的一个R包,灵感来源于pheatmap,因此有很多与pheatmap类似的参数。
如果你使用了ComplexHeatmap,那记得引用文献:
“Gu, Z. (2016) Complex
heatmaps reveal patterns and correlations in multidimensional genomic
data. DOI: 10.1093/bioinformatics/btw313”。
而本系列推文所包含的内容和数据均学习自ComplexHeatmap使用文档,并根据我自己的理解进行阐述,代码部分根据我所接触的打赏营学员的一些习惯进行了通俗化的调整。好了,接下来我将从易到难,给大家展示神奇的ComplexHeatmap包的使用方法:
#安装并加载ComplexHeatmap包 #方法一 #library(devtools) #install_github( "jokergoo/ComplexHeatmap") #方法二 #if(!requireNamespace( "BiocManager", quietly = TRUE)) #install.packages( "BiocManager") #BiocManager::install( "ComplexHeatmap") library(ComplexHeatmap) library(circlize)
常规热图
作为一个最全面的R包,用ComplexHeatmap包当然也可以用来绘制类似于ggplot2和pheatmap等包绘制的常规热图,这也是一个热图最主要的载体。首先让我们生成一个随机矩阵,其中按列分为三组,按行分为三组:
set.seed(123) nr1 = 4; nr2 = 8; nr3 = 6; nr = nr1 + nr2 + nr3 nc1 = 6; nc2 = 8; nc3 = 10; nc = nc1 + nc2 + nc3 #以matrix(rnorm(nr1*nc1, mean = 1, sd = 0.5), nr =
nr1)为例,表示使用rnorm函数构建一个正态分布,nr1*nc1=4*6=24表示这个正态分布有24个值,均数为1,标准差为0.5,然后使用matrix函数构建一个矩阵,函数为nr1也就是4行;下面每一行的构建都为同一个意思,然后使用rbind根据行进行叠加,使用cbind将3个rbind的结果进行列的叠加,这样就构建了一个18行24列的矩阵 mat <- cbind(rbind(matrix(rnorm(nr1*nc1, mean = 1, sd = 0.5), nr = nr1), matrix(rnorm(nr2*nc1, mean = 0, sd = 0.5), nr = nr2), matrix(rnorm(nr3*nc1, mean = 0, sd = 0.5), nr = nr3)), rbind(matrix(rnorm(nr1*nc2, mean = 0, sd = 0.5), nr = nr1), matrix(rnorm(nr2*nc2, mean = 1, sd = 0.5), nr = nr2), matrix(rnorm(nr3*nc2, mean = 0, sd = 0.5), nr = nr3)), rbind(matrix(rnorm(nr1*nc3, mean = 0.5, sd = 0.5), nr = nr1), matrix(rnorm(nr2*nc3, mean = 0.5, sd = 0.5), nr = nr2), matrix(rnorm(nr3*nc3, mean = 1, sd = 0.5), nr = nr3))) #使用sample函数进行随机抽样,在这里的作用其实就是将前面构建的mat的数据打散 mat <- mat[sample(nr, nr), sample(nc, nc)] rownames(mat) = paste0("gene", seq_len(nr)) colnames(mat) = paste0("Sample", seq_len(nc)) head(mat)## Sample1 Sample2 Sample3 Sample4 Sample5 Sample6 ## gene1 0.90474160 -0.35229823 0.5016096 1.26769942 0.8251229 0.16215217 ## gene2 0.90882972 0.79157121 1.0726316 0.01299521 0.1391978 0.46833693 ## gene3 0.28074668 0.02987497 0.7052595 1.21514235 0.1747267 0.20949120 ## gene4 0.02729558 0.75810969 0.5333504 -0.49637424 -0.5261114 0.56724357 ## gene5 -0.32552445 1.03264652 1.1249573 0.66695147 0.4490584 1.04236865 ## gene6 0.58403269 -0.47373731 0.5452483 0.86824798 -0.1976372 -0.03565404 ## Sample7 Sample8 Sample9 Sample10 Sample11 Sample12 ## gene1 -0.2869867 0.68032622 -0.1629658 0.8254537 0.7821773 -0.49625358 ## gene2 1.2814948 0.38998256 -0.3473535 1.3508922 1.1183375 2.05005447 ## gene3 -0.6423579 -0.31395304 0.2175907 -0.2973086 0.4322058 -0.25803192 ## gene4 0.8127096 -0.01427338 1.0844780 0.2426662 0.8783874 1.38452112 ## gene5 2.6205200 0.75823530 -0.2333277 1.3439584 0.8517620 0.85980233 ## gene6 -0.3203530 1.05534136 0.7771690 0.4594983 0.2550648 -0.02778098 ## Sample13 Sample14 Sample15 Sample16 Sample17 Sample18 ## gene1 -0.0895258 -0.35520328 0.1072694 0.96322199 -0.39245223 -0.1878014 ## gene2 1.3770269 -0.77437640 0.9829664 0.23854381 -0.53589561 1.3003544 ## gene3 -0.5686518 -0.51321045 -0.0451598 0.82272880 -0.02251386 0.2427300 ## gene4 0.8376570 0.10797078 0.4520019 0.81648036 0.02650211 0.4072600 ## gene5 1.9986067 -0.50928769 0.7708173 -0.09421702 -1.15458444 1.2715970 ## gene6 -0.2112484 0.01669142 -0.3259750 0.54460361 0.89101254 0.3699738 ## Sample19 Sample20 Sample21 Sample22 Sample23 Sample24 ## gene1 1.08736320 0.7132199 -0.1853300 -0.14238650 0.6407669 1.3266288 ## gene2 0.14237891 0.4471643 0.4475628 -0.31251963 0.7057150 0.8120937 ## gene3 1.09511516 0.5852612 0.1926402 0.51278568 1.6361334 1.1339175 ## gene4 -0.02625664 0.9556956 0.3443201 0.07668656 1.7857291 0.5280084 ## gene5 0.60599022 0.8304101 -0.1902355 -0.14753574 1.0123366 0.2140750 ## gene6 0.81537706 1.2737905 1.8575325 0.88491126 0.5670193 0.5372756
在使用自己的数据进行绘图时候并不需要上面这么复杂,只需要把你的数据整理为mat一样的行列格式,导入后转换为matrix即可(as.matrix)。当你有了这样一个矩阵之后,使用ComplexHeatmap绘制热图就跟使用pheatmap绘制热图一样简单:
ht<- Heatmap(mat) draw(ht)
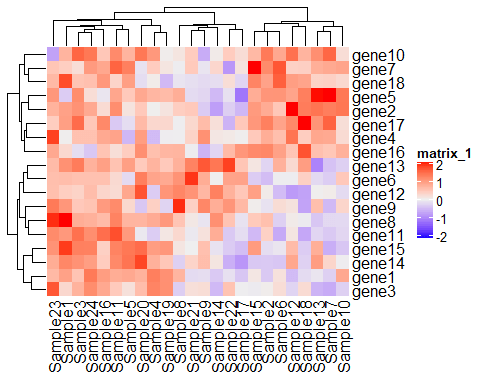
使用mat作为热图名称,可以方便后续对这个热图叠加参数,在打赏营我说过,可视化的过程无非就是“拆”和“叠”,在用ComplexHeatmap绘制复杂热图时候也很好体现了这一点,我们接着对这个热图进行一定的美化:
# 首先是颜色 col_fun <- colorRamp2(c(- 2, 0, 2), c( "navy", "black", "yellow")) col_fun(se q(-3, 3))Heatmap(mat, name = "mat", col = col_fun)
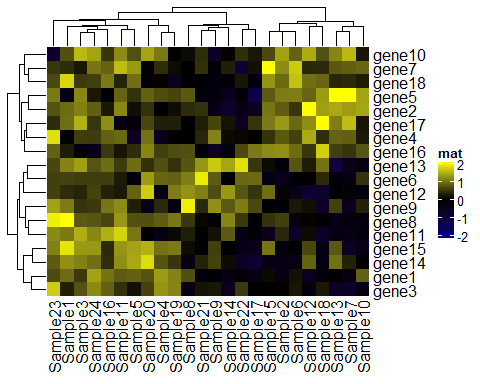
设置相同的颜色可以使数据之间更具有可比性,假设你有三个数据集,那么你只需要绘制三个热图:
Heatmap(mat, name = "mat", col = col_fun, column_title = "mat")
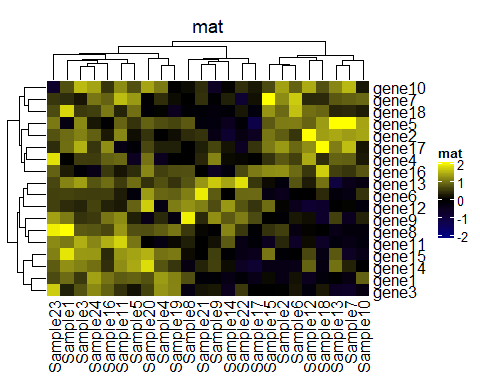
Heatmap(mat/ 4, name = "mat", col = col_fun, column_title = "mat/4")

Heatmap( abs(mat), name = "mat", col = col_fun, column_title = "abs(mat)")

ComplexHeatmap手册写到这种设置颜色的方法并不会受到离群值的影响,此外,你也可以使用连续型向量作为颜色映射,但是可能受到离群值的影响,我们来比较一下两种方法:
mat2 < -mat # 添加一个离群值 mat2[ 1, 1] = 100000 Heatmap( mat2, name= "mat", col= col_fun)
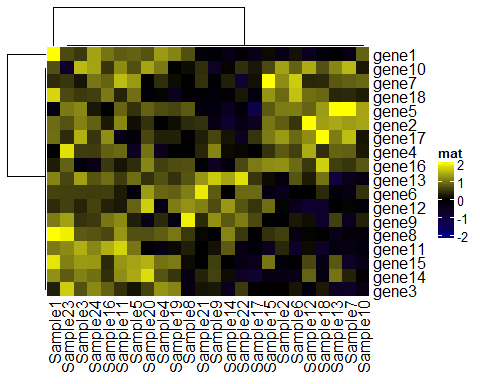
Heatmap(mat2, name = "mat", col = rev(rainbow( 10)))

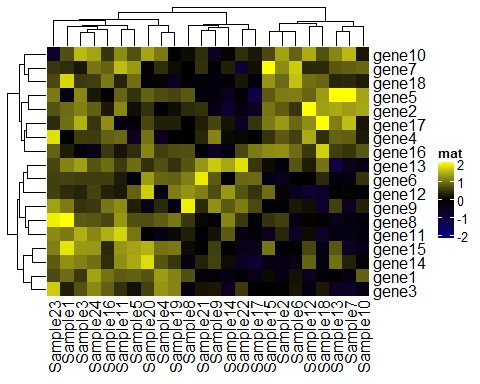
# 没有离群值的情况 Heatmap(mat, name = "mat", col = col_fun)
Heatmap(mat, name = "mat", col = rev(rainbow( 10)))
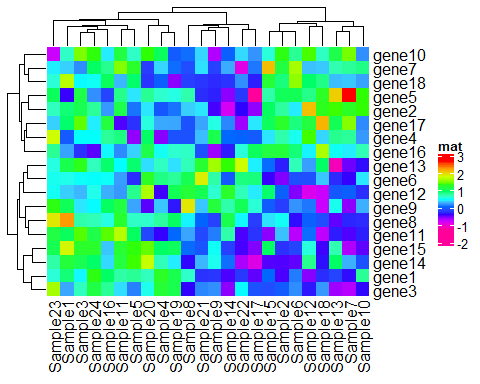
比较上面四个图我们发现1图和3图的结果一致,而2图仅仅因为100000的影响,就会导致整体颜色受到偏差。这是因为我们设置连续型向量进行颜色映射时候,因为映射从矩阵的最小值开始,以最大值结束,有一个离散值的存在,则会把其他值都推向另一个方向呈现同一种颜色,所以以后你在绘制热图时出现这种情况,可以先考虑下是不是你的颜色映射出了问题。
当你的数据有NA值的时候,也一样可以绘制热图,我们来试试:
mat_with_na <- mat # prob即probability,表示取随机数的概率 na_index <- sample(c( TRUE, FALSE), nrow(mat)*ncol(mat), replace = TRUE, prob = c( 1, 9)) # 将TRUE的地方变为NA mat_with_na[na_index] <- NA # 使用na_col将NA值的地方变为黑色 Heatmap(mat_with_na, name = "mat", na_col = "black")
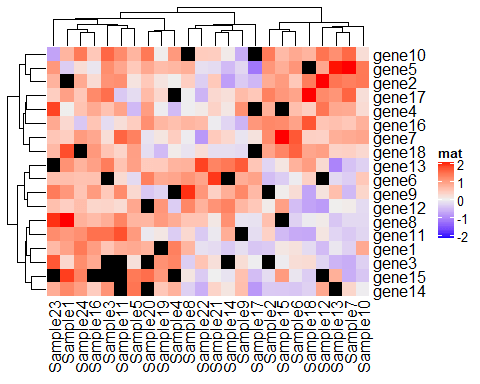
此外,热图边界的颜色可以通过border和rect gp参数设置。border控制热图主体的全局边界,rect gp控制热图中网格的边界:
Heatmap(mat, name = "mat", border = "black", rect_gp = grid::gpar(col = "white", lwd = 3))
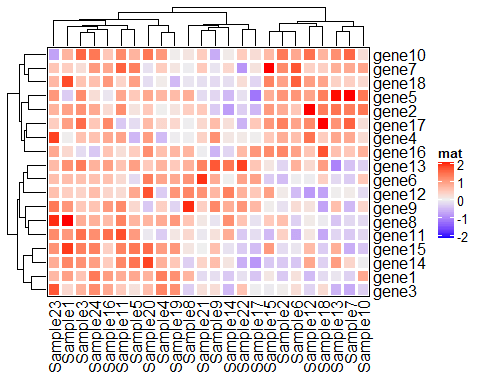
Heatmap(mat, name = "mat", rect_gp = grid::gpar(col= 1: 10,lwd = 3))
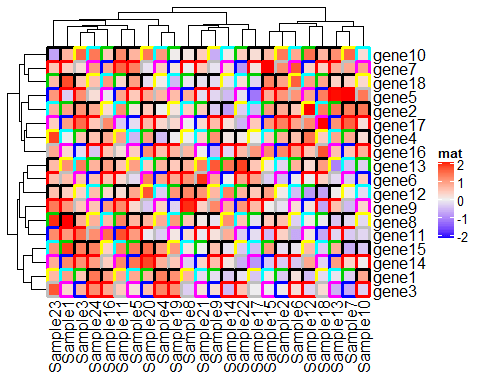
Heatmap(mat, name = "mat", rect_gp = grid::gpar( type= "none"))
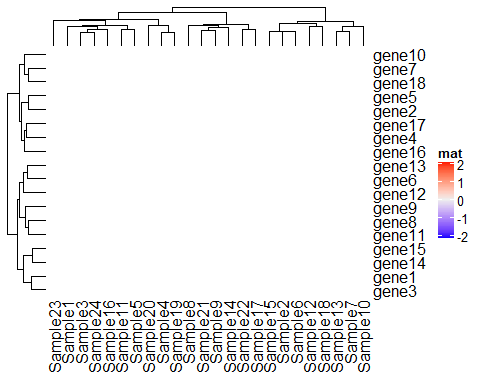
当我们把type设置为none的时候,整个热图主体界面就变成了空白,这时候我们可以在这个空间任意发挥,在后面对热图进行修饰时候我们还会多次用到这个参数。 作为一个顶级的绘图包,当然也可以给热图加上标题:
Heatmap(mat, name = "mat", column_title = "用我标注列名", row_title = "用我标注行名")
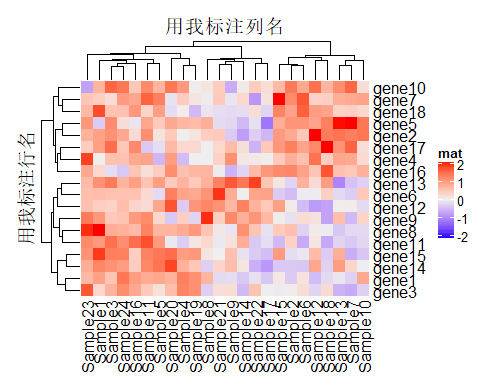
Heatmap(mat, name = "mat", column_title = "用我标注列名", column_title_side = "bottom", #把列名放在底部 column_title_gp = gpar(fontsize = 20, fontface = "bold"), row_title = "用我标注行名")
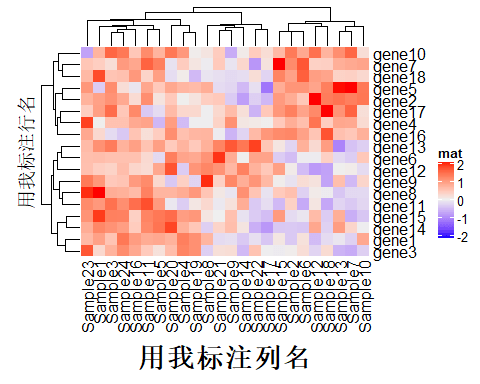
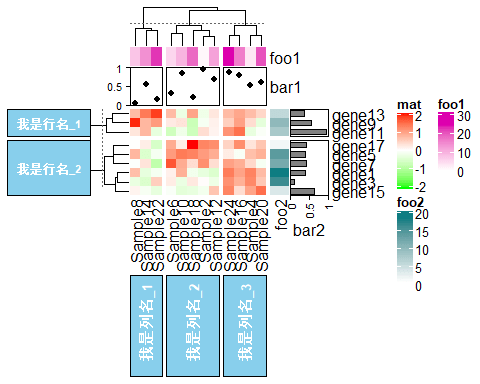
# 花样设置,可以对热图的聚类进行切割,方便看出聚类结果 Heatmap(mat, name= "mat", column_km= 3, column_title= "我是列名_%s", column_title_rot= 90, column_title_side= "bottom", #把列名放在底部 column_title_gp= gpar(fontsize = 10, fontface= "bold", fill= "skyblue", col= "white", border= "black"), row_title_rot= 0, #行名转变为平行 row_km= 2, row_title= "我是行名_%s", row_title_gp= gpar(fontsize = 8, fontface= "bold", fill= "skyblue", col= "white", border= "black"))
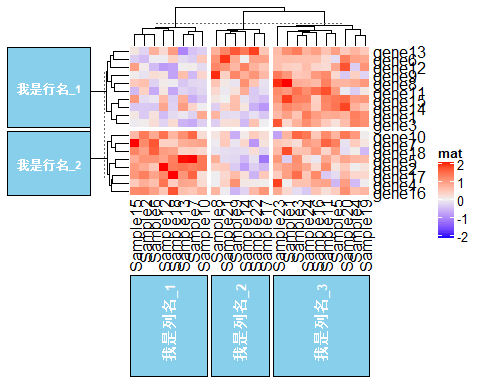
这样的热图,你只需要在聚类后把相应的行名和列名的特征打上去,就显得比别人常规的热图高级一步啦,比如行名可以是上下调,也可以是T细胞相关markers或者B细胞相关markers等等,列名可以是正常or肿瘤,或者T1 or T2 or T3 等等。
好啦,今天我们先到这里,下次我们继续对这个热图进行修饰,我们的目的是以一个图片为引子,通过不断叠加,学会一个R包并且用于文章之中,我这里再卖一个关子,ComplexHeatmap可不止能画热图而已哦O(∩_∩)O 我们下回见!

