考虑到很多做实验的小伙伴对很多生物信息学概念不是很了解,受实验小白的委托,我给大家写了一个非常简单的工具:KEGG富集分析
KEGG是干嘛的捏?
我这么跟你说吧:人类的七千多个基因组都是有已知功能的,KEGG把这七千多个基因分成了300个类,就是我们通常说的kegg通路;比如,我现在做了个实验,发现某细胞系里面的两万个基因里面有300个基因变化了,那这300个基因会涉及到KEGG数据库的哪几个通路?这时候就需要用到我的工具啦~将这300个基因加入工具里面,得出结果:有30个Cell cycle通路。
就这么简单?可是富集分析怎么分析呐?
接下来还会用到我这个小工具:事实上,Cell Cycle KEGG 通路 hsa04110只有124个基因;而我处理了细胞系之后,在只有300个基因发生统计学显著变化的情况下,就有30个是Cell Cycle通路?高达10%的概率,那到底这个Cell Cycle通路是不是被显著改变了呢? 首先,我会把用户的300个基因,都用KEGG数据库的300个通路注释,然后一个个通路循环做超几何分布检验,给出P值;比如刚才的Cell Cycle 通路就很显著,因为124/7000就2%的概率,结果我 30/300有10%的概率~太可怕了,所以我的这个处理显著的改变了细胞系的Cell Cycle 通路。
使用方法其实没什么好说的,就是复制粘贴你感兴趣的基因到输入框即可。我这里主要讲解,这个网页如何写出来的。
一、代码编写
UI界面的代码:
-
suppressPackageStartupMessages(library(shiny))
-
suppressPackageStartupMessages(library(ggplot2))
-
suppressPackageStartupMessages(library(DT))
-
suppressPackageStartupMessages(library(stringr) )
-
suppressPackageStartupMessages(library(clusterProfiler))
-
suppressPackageStartupMessages(library(shinyjs))
-
suppressPackageStartupMessages(library(shinyBS))
-
## 这个是侧边栏,就是你看到的网页坐标的输入框
-
## 分别是 下拉框,文字输入框,多选按钮,确定按钮。
-
sp <- sidebarPanel(
-
selectInput("IDtype",
-
label = "choose the ID type",
-
choices = c("HUGO Gene Symbol"= "symbols",
-
"Entrez Gene ID"= "geneIds",
-
"ENSEMBL Gene ID"= "geneEnsembl")),
-
## "symbols" "geneIds" "geneNames" "geneEnsembl" "geneMap" "geneAlias"
-
h3("Type the Gene List:"),
-
tags$style(type="text/css", "textarea {width:100%}"),
-
tags$textarea(id = 'input_text',
-
placeholder = "TP53nCBX6",
-
rows = 8, ''),
-
hr(),
-
radioButtons("species", "Select a species:",
-
c("human(Homo sapiens)"="human",
-
"mouse(Mus Musculus)"="mouse"),
-
selected = 'human'
-
),
-
hr(),
-
bsAlert("alert_ui_anchorId"),
-
actionButton("do", "analysis", icon("paper-plane"),
-
style="color: #fff; background-color: #337ab7; border-color: #2e6da4"
-
)
-
)
-
## 这个是主栏,就显示两个表格即可。
-
mp <- mainPanel(
-
h3('KEGG enrichment:'),
-
DT::dataTableOutput('KEGG_df'),
-
hr() ,
-
h3('Gene Annotation result:'),
-
DT::dataTableOutput('gene_df'),
-
hr()
-
)
-
## 主程序入口,是一个侧边栏格式的网页,所以需要定义侧栏和主栏!
-
shinyUI(
-
fluidPage(
-
titlePanel('KEGG enrichment'),
-
sidebarLayout(
-
sidebarPanel = sp,
-
mainPanel = mp
-
)
-
)
-
)
server服务端代码
-
suppressPackageStartupMessages(library(shiny))
-
suppressPackageStartupMessages(library(ggplot2))
-
suppressPackageStartupMessages(library(DT))
-
suppressPackageStartupMessages(library(stringr) )
-
suppressPackageStartupMessages(library(clusterProfiler))
-
suppressPackageStartupMessages(library(shinyjs))
-
suppressPackageStartupMessages(library(shinyBS))
-
suppressMessages(library(RSQLite))
-
sqlite <- dbDriver("SQLite")
-
createLink <- function(base,val) {
-
sprintf('<a href="%s" class="btn btn-link" target="_blank" >%s</a>',base,val) ##target="_blank"
-
}
-
log_cat <- function(info='hello world~',file='log.txt'){
-
cat(as.character(Sys.time()),info ,"n",file=file,append=TRUE)
-
}
-
shinyServer(function(input, output,session) {
-
## 定义几个全局变量
-
glob_values <- reactiveValues(
-
gene_df=NULL,
-
kegg_df=NULL,
-
species=NULL
-
)
-
reactiveValues.reset <-function(){
-
glob_values$gene_df=NULL
-
glob_values$species=NULL
-
glob_values$kegg_df=NULL
-
}
-
## 主程序入口,用户点击了确定运行程序的按钮,就开始判断文字输入框是否有基因列表。
-
observeEvent(input$do,{
-
reactiveValues.reset()
-
db=ifelse(input$species == 'human','human_gene_info','mouse_gene_info')
-
gene_list=NULL
-
## first check the upload file:
-
inFile <- input$file1
-
if( ! is.null(inFile) ){
-
gene_list=read.table(inFile$datapath, header=input$header)[,1]
-
}
-
## Then check the text input area:
-
if( nchar(input$input_text) >5){
-
gene_list = strsplit(input$input_text,'n')[[1]]
-
}
-
if( !is.null(gene_list)){
-
if(length(gene_list) < 20){
-
createAlert(session, "alert_ui_anchorId", "exampleAlert", title = "Oops",
-
content = " 你给的基因数量少于20,没啥意思,不给你富集了 !", append = FALSE)
-
}elseif(length(gene_list) > 2000){
-
createAlert(session, "alert_ui_anchorId", "exampleAlert", title = "Oops",
-
content = " 给我多于2000个基因,我的服务器受不了呀,要不你先捐赠一下呗 ", append = FALSE)
-
}else{
-
closeAlert(session, "exampleAlert")
-
gene_list=unique(gene_list)
-
con <- dbConnect(sqlite,"hg19_bioconductor.sqlite")
-
sql <- paste0('select * from ',db," where ",input$IDtype,
-
" in ('",paste0(gene_list,collapse = "','"),"')")
-
glob_values$gene_df=dbGetQuery(con, sql)
-
dbDisconnect(con)
-
if(T ){ ## for kegg
-
suppressPackageStartupMessages(library(org.Mm.eg.db))
-
suppressPackageStartupMessages(library(org.Hs.eg.db))
-
gene_df = glob_values$gene_df
-
############################################################
-
############ gene ID transfer #############################
-
############################################################
-
gene_list <- gene_df$symbol
-
gene.df=''
-
if(input$species == 'human'){
-
gene.df <- bitr(gene_list, fromType = "SYMBOL",
-
toType = c("ENSEMBL", "ENTREZID"),
-
OrgDb= org.Hs.eg.db )
-
ego <- enrichKEGG(gene = gene.df$ENTREZID,
-
organism = 'hsa',
-
pvalueCutoff = 0.99,
-
qvalueCutoff=0.99
-
)
-
ego <- setReadable(ego, OrgDb= org.Hs.eg.db, keytype="ENTREZID")
-
}else{
-
gene.df <- bitr(gene_list, fromType = "SYMBOL",
-
toType = c("ENSEMBL", "ENTREZID"),
-
OrgDb= org.Mm.eg.db )
-
ego <- enrichKEGG(gene = gene.df$ENTREZID,
-
organism = 'mmu',
-
pvalueCutoff = 0.99,
-
qvalueCutoff=0.99
-
)
-
ego <- setReadable(ego, OrgDb= org.Mm.eg.db, keytype="ENTREZID")
-
}
-
glob_values$kegg_df <- as.data.frame(ego)
-
} ## for kegg
-
}} ## if( !is.null(gene_list)){
-
})
-
## 显示第一个表格,基因注释表格
-
output$gene_df <- DT::renderDataTable({
-
if(! is.null(glob_values$gene_df)){
-
dat=glob_values$gene_df
-
dat$geneIds=createLink(paste0("http://www.ncbi.nlm.nih.gov/gene/",dat$geneIds),dat$geneIds)
-
dat
-
}
-
} ,rownames= FALSE,escape = FALSE,options = list(
-
pageLength = 10,
-
lengthMenu = list(c(10, 50, 100,-1), c('10', '50','100', 'All'))
-
)## endforoptions
-
)
-
## 显示第二个表格,kegg富集结果表格
-
output$KEGG_df <- DT::renderDataTable({
-
if(! is.null(glob_values$kegg_df))
-
glob_values$kegg_df
-
} ,rownames= FALSE,options = list(
-
pageLength = 10,
-
lengthMenu = list(c(10, 50, 100,-1), c('10', '50','100', 'All'))
-
,
-
scrollX = TRUE,
-
fixedHeader = TRUE,
-
fixedColumns = TRUE ,
-
deferRender = TRUE
-
),
-
#filter = 'top',
-
escape = FALSE
-
)
-
})
组合两个程序,在Rstudio里面运行即可
二、安装及运行
1、首先安装R,再安装Rstudio,然后安装shiny等R包。
2、把上面UI端代码拷贝成ui.R 的文件,再把服务端代码拷贝成server.R文件,放在同一个文件夹,命名为yourAPP里面。
3、进入R里面,该文件夹上层目录,用runAPP('yourAPP')来执行即可
直通车寄语
当然,最简单的就是去https://github.com/jmzeng1314/myShiny 我的GitHub里面把代码下载,或者查看其它工具咯
UI界面如下:
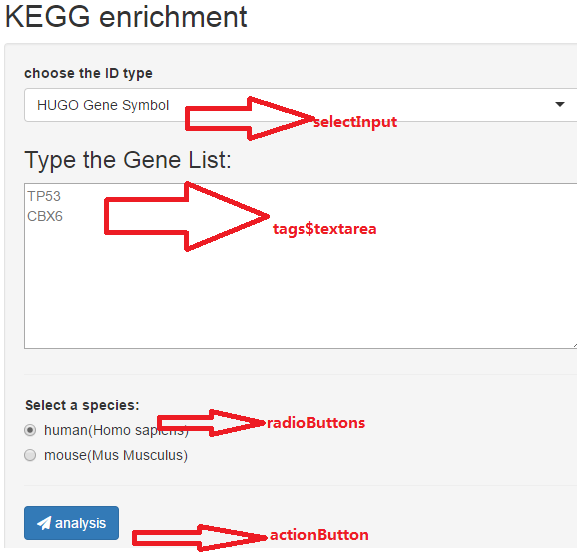
点击阅读原文,试用小工具,赶快输入一串基因去调戏一下这个工具吧!
比如:
LGALS1
DUSP10
LOC402644
SOX11
LOC100131940
LSM5
HEATR2
UCP2
ASNS
RNU6-15
RNU6-1
DUSP10
CDCA7L
DBNL
AXL
TXNRD2
HNRNPA2B1
DHRS2
- 本文固定链接: https://maimengkong.com/kyjc/1066.html
- 转载请注明: : 萌小白 2022年6月29日 于 卖萌控的博客 发表
- 百度已收录
