Pathview是一个可视化KEGG通路的R包,它会在线从KEGG网站上下载KEGG通路,并进行个性化处理,例如填上不同的颜色 。 今天就来分享一下如何用Pathview画出高大上的基因与代谢通路热图。 基因热通路图
在此之前现在R中把必要的包安装好,并加载上
if (!requireNamespace("BiocManager", quietly=TRUE)) install.packages("BiocManager") BiocManager::install(c("Rgraphviz", "png", "KEGGgraph", "org.Hs.eg.db","pathview","gage")) library(pathview) 首先,我们把数据准备好,第一列为基因的ENTREZ号,第二列为基因的变化倍数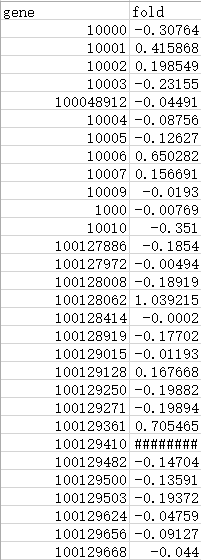
把数据导入R中data<-read.csv("data1.xls",row.names=1,sep="\t",head=T)
head(data,10)
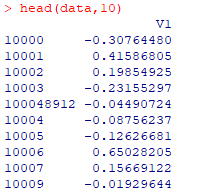
开始进行绘制图片,gene.data是用于绘图的数据,pathway.id是需要绘制的KEGG通路号,species是物种的简称,out.suffix是导出的数据后缀pv.out <- pathview(gene.data = data, pathway.id = "04110",species = "hsa", out.suffix = "data1")
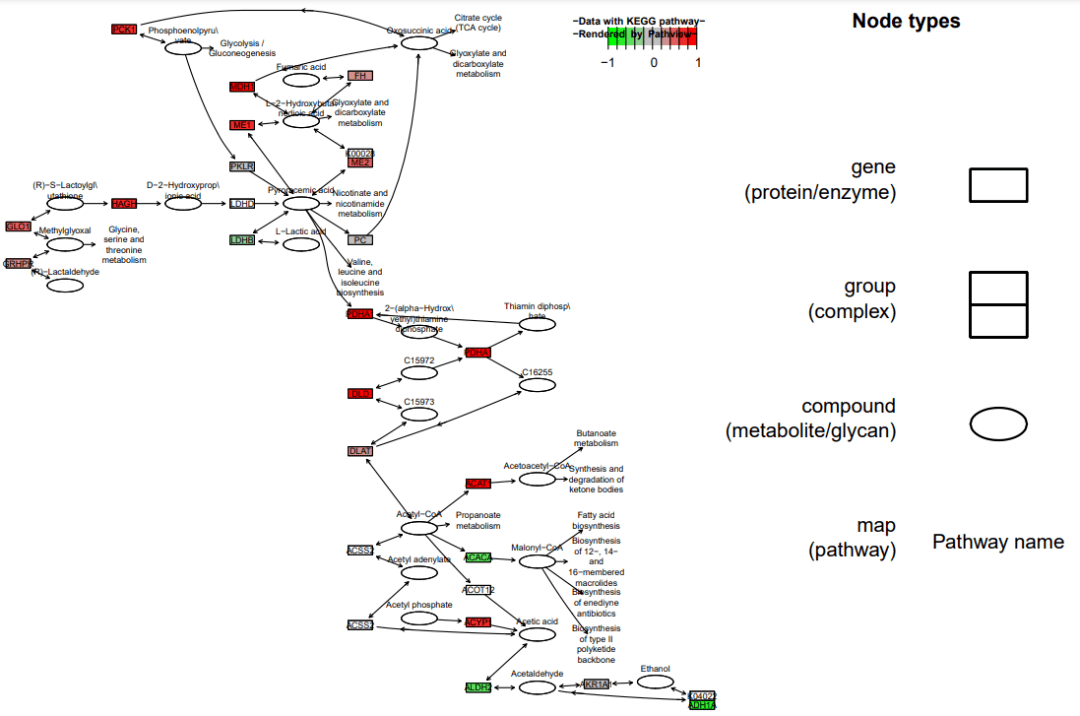
运行完成后,可以看到文件夹下出现了三个文件,其中带data1的是绘制完的图片,其他是用于绘图的KEGG原始数据。 查看图片结果,可以看到基因的变化已经用热图的形式展现在通路上了
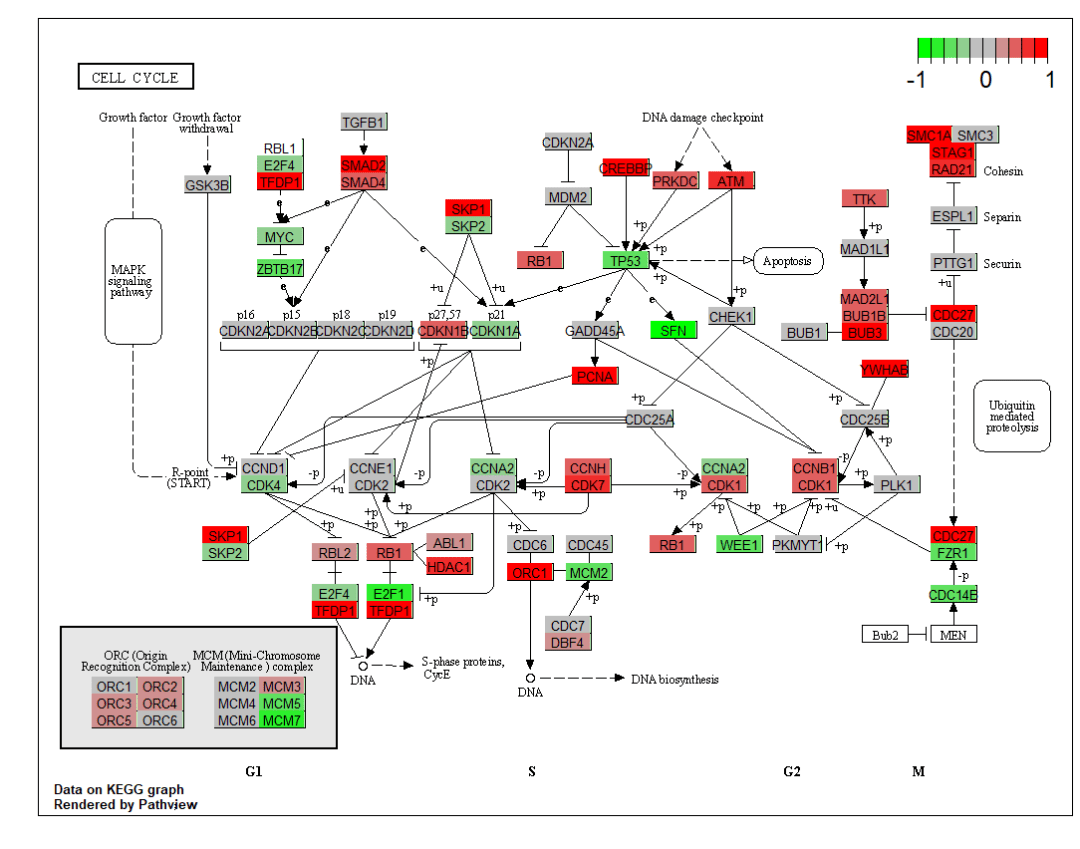
查看图片结果,可以看到基因的变化已经用热图的形式展现在通路上了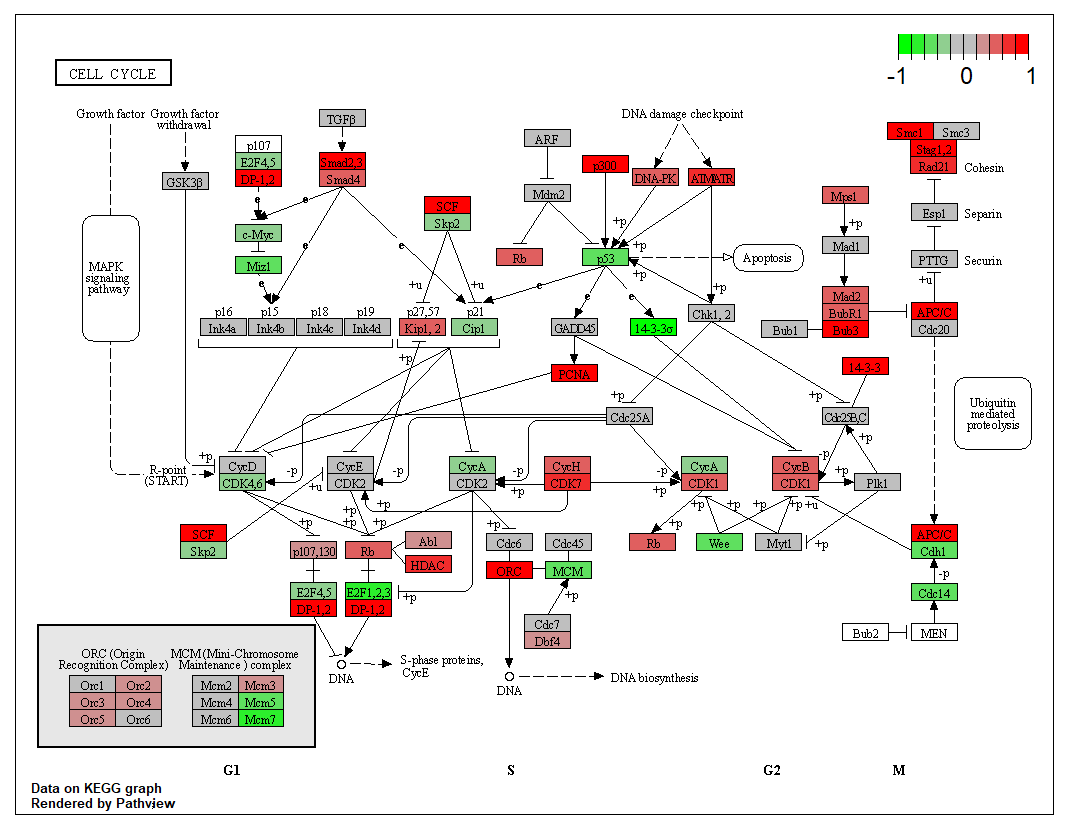 分为两个图层,图像的立体感增加了pv.out <- pathview(gene.data = data, pathway.id = demo.paths$sel.paths[i],
分为两个图层,图像的立体感增加了pv.out <- pathview(gene.data = data, pathway.id = demo.paths$sel.paths[i],
species = "hsa", out.suffix = "data1", kegg.native = T,
same.layer = F)
 查看物种所有通路 paths.hsa
查看物种所有通路 paths.hsa
 也可以批量对几条通路同时进行上色,先定义几条通路
也可以批量对几条通路同时进行上色,先定义几条通路 得到了所有选择的通路结果
得到了所有选择的通路结果 可以直接导出pdf格式,先定义图例位置
可以直接导出pdf格式,先定义图例位置 pv.out <- pathview(gene.data = data, pathway.id = mypath,
pv.out <- pathview(gene.data = data, pathway.id = mypath,
species = "hsa", out.suffix = "data3", kegg.native = F,
sign.pos = sign)定义图例位置
 不同反应分开画 pv.out
<- pathview(gene.data = data, pathway.id = demo.paths$sel.paths[i],
species = "hsa", out.suffix = "data4", kegg.native = F, sign.pos =
demo.paths$spos[i], split.group = T)
不同反应分开画 pv.out
<- pathview(gene.data = data, pathway.id = demo.paths$sel.paths[i],
species = "hsa", out.suffix = "data4", kegg.native = F, sign.pos =
demo.paths$spos[i], split.group = T) 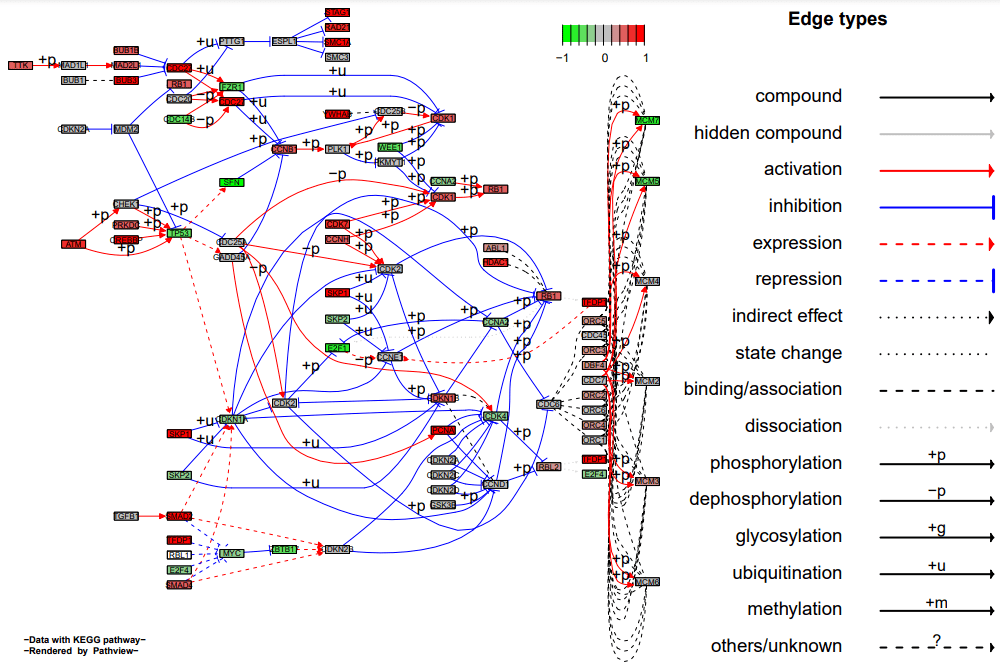
线段拓宽pv.out <- pathview(gene.data = data, pathway.id = demo.paths$sel.paths[i],
species = "hsa", out.suffix = "data5", kegg.native = F,
sign.pos = demo.paths$spos[i], split.group = T, expand.node = T)
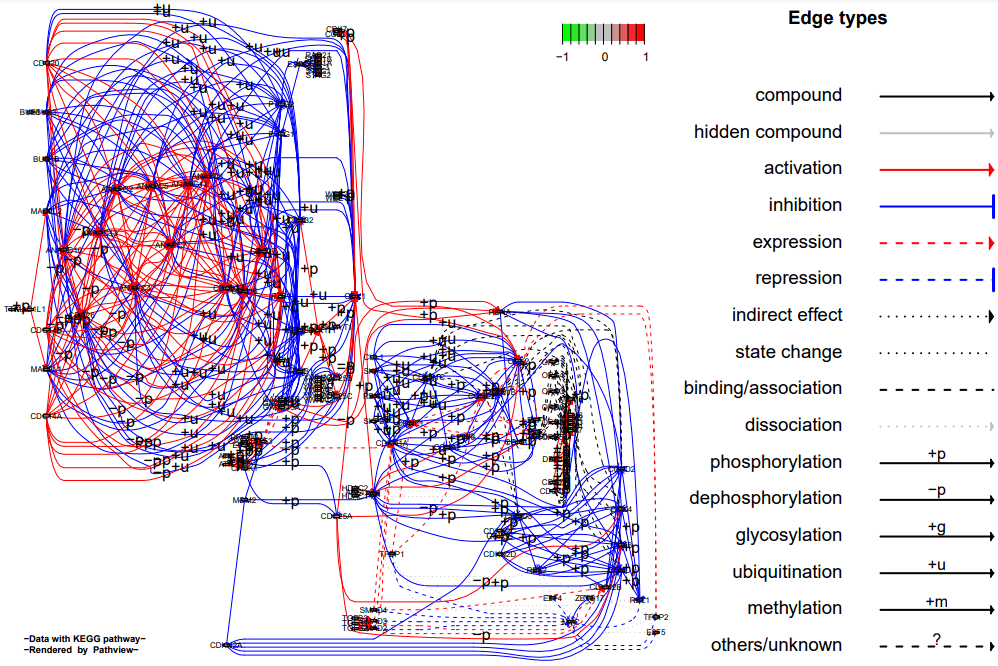 代谢物热通路图
代谢物热通路图
下面开始画代谢物的通路热图,第一列是化合物的CPD号,第二列是变化倍数 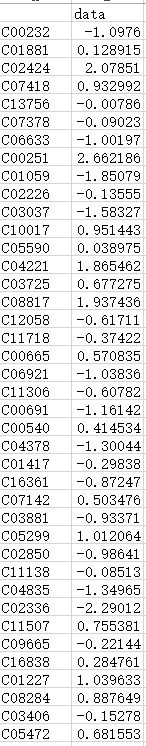 导入数据,并开始绘制 data2<-read.csv("data2.xls",row.names=1,sep="\t",head=T)
导入数据,并开始绘制 data2<-read.csv("data2.xls",row.names=1,sep="\t",head=T)
pv.out <- pathview(cpd.data = data2,
pathway.id = demo.paths$sel.paths[i], species = "hsa", out.suffix = "data6",
keys.align = "y", kegg.native = T, key.pos = demo.paths$kpos1[i])
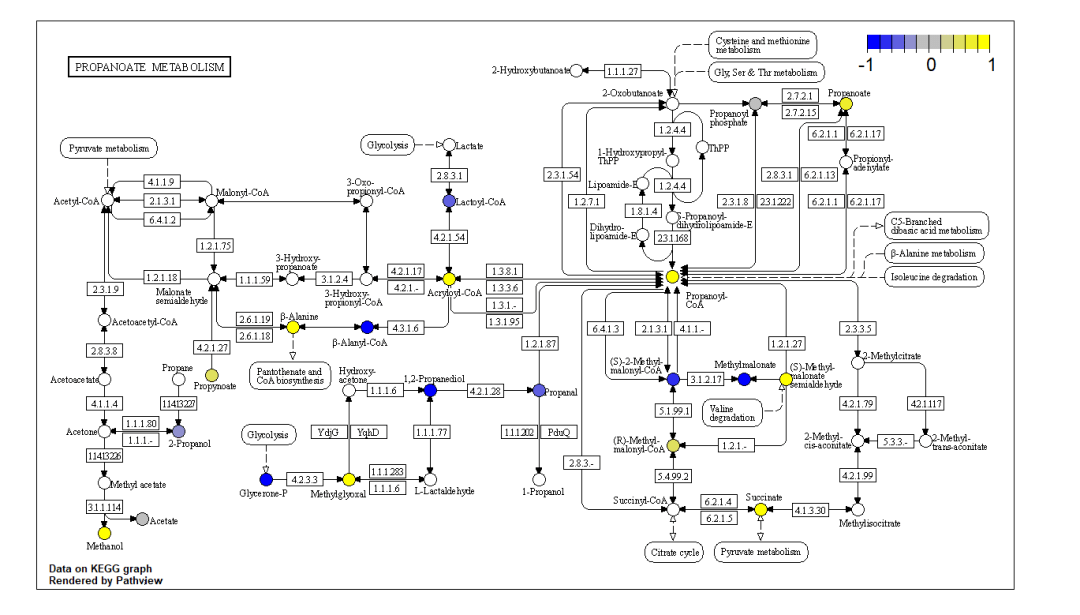 同时绘制基因与代谢物通路热图 pv.out
<- pathview(gene.data = data, cpd.data = data2, pathway.id =
demo.paths$sel.paths[i], species = "hsa", out.suffix = "data7",
keys.align = "y", kegg.native = F, key.pos = demo.paths$kpos2[i],
sign.pos = demo.paths$spos[i], cpd.lab.offset = demo.paths$offs[i])
同时绘制基因与代谢物通路热图 pv.out
<- pathview(gene.data = data, cpd.data = data2, pathway.id =
demo.paths$sel.paths[i], species = "hsa", out.suffix = "data7",
keys.align = "y", kegg.native = F, key.pos = demo.paths$kpos2[i],
sign.pos = demo.paths$spos[i], cpd.lab.offset = demo.paths$offs[i]) 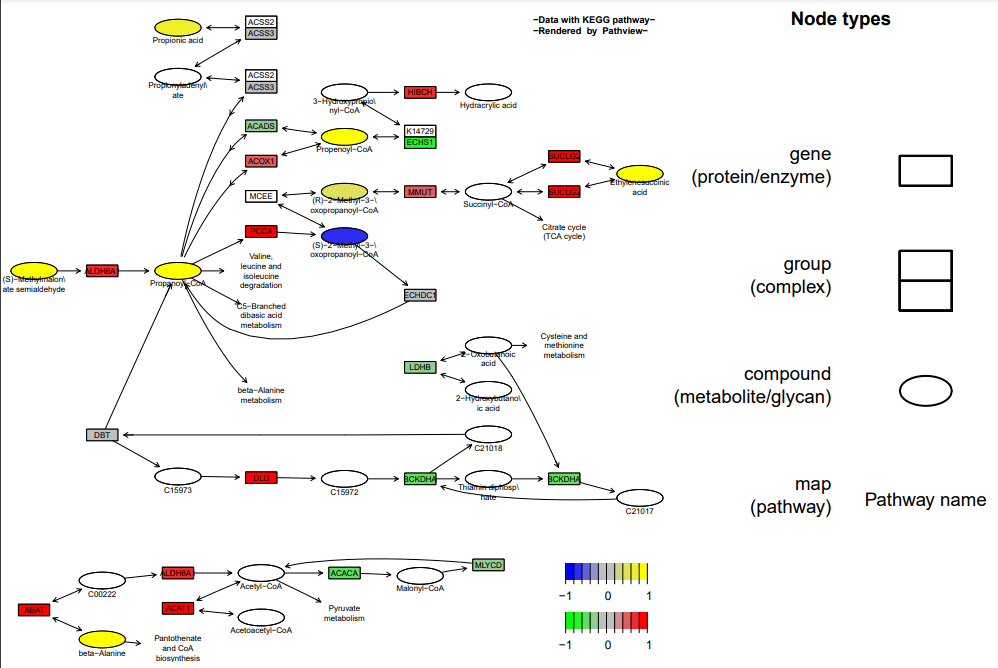
展现不同数据中同一条通路的代谢物变化,先准备数据,并导入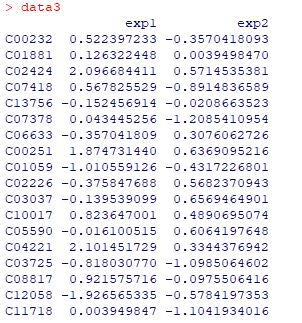
pv.out <- pathview(gene.data = gse16873.d[, 1:3],
cpd.data = data3, pathway.id = demo.paths$sel.paths[i],
species = "hsa", out.suffix = "data8", keys.align = "y",
kegg.native = T, match.data = F, multi.state = T, same.layer = T)
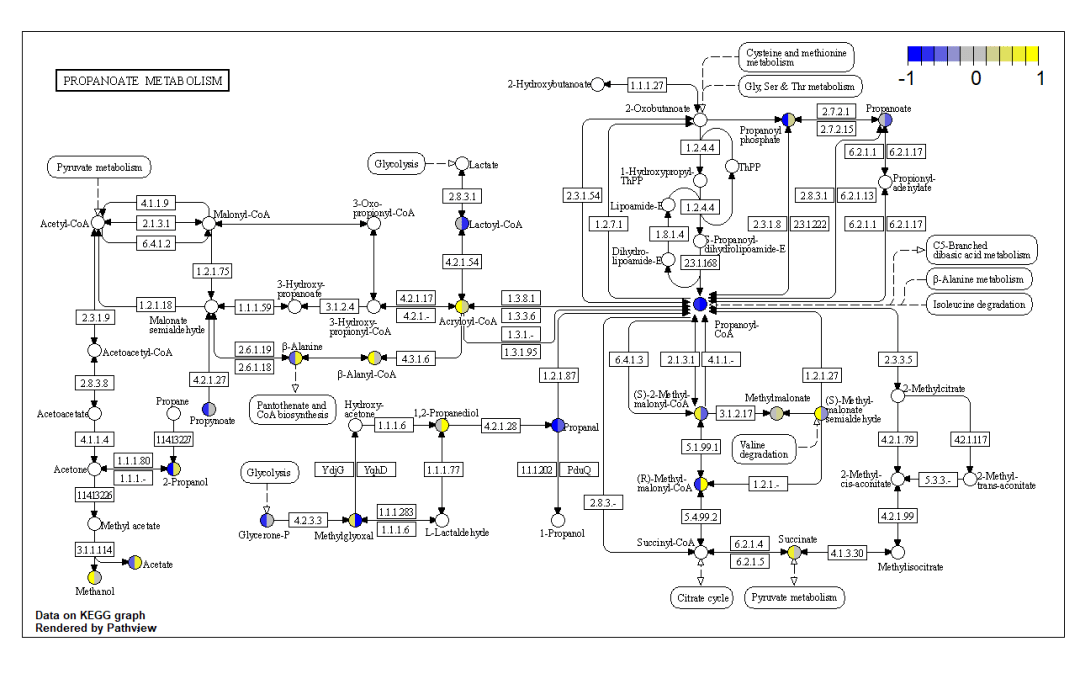 导出pdf结果 pv.out
<- pathview(gene.data = gse16873.d[, 1:3], cpd.data =
sim.cpd.data2[, 1:2], pathway.id = demo.paths$sel.paths[i], species =
"hsa", out.suffix = "data9", keys.align = "y", kegg.native = F,
match.data = F, multi.state = T, same.layer = T, key.pos =
demo.paths$kpos2[i], sign.pos = demo.paths$spos[i])
导出pdf结果 pv.out
<- pathview(gene.data = gse16873.d[, 1:3], cpd.data =
sim.cpd.data2[, 1:2], pathway.id = demo.paths$sel.paths[i], species =
"hsa", out.suffix = "data9", keys.align = "y", kegg.native = F,
match.data = F, multi.state = T, same.layer = T, key.pos =
demo.paths$kpos2[i], sign.pos = demo.paths$spos[i])
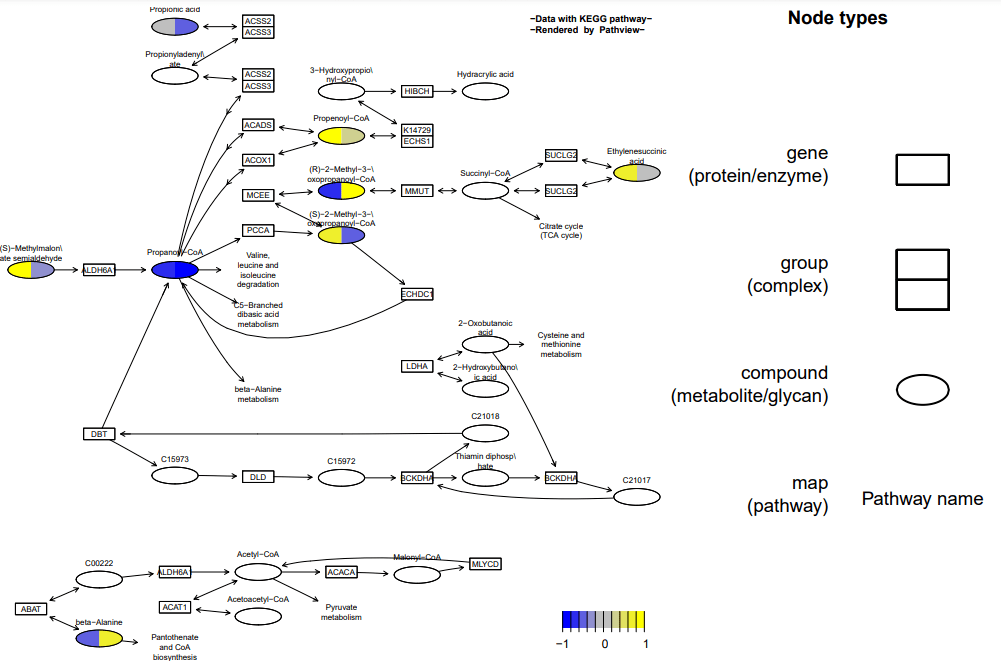 准备多组数据并导入
准备多组数据并导入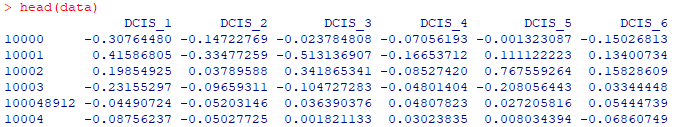 进行t检验data.t <- apply(data, 1, function(x) t.test(x,
进行t检验data.t <- apply(data, 1, function(x) t.test(x,
alternative = "two.sided")$p.value)
根据基因的t检验结果和代谢物的差异倍数进行筛选
sel.genes <- names(data.t)[data.t < 0.1]
sel.cpds <- names(sim.cpd.data)[abs(sim.cpd.data) > 0.5]
得到挑选的基因和代谢物的结果 pv.out <- pathview(gene.data = sel.genes, cpd.data = sel.cpds, pathway.id = demo.paths$sel.paths[i], species = "hsa", out.suffix = "sel.genes.sel.cpd", keys.align = "y", kegg.native = T, key.pos = demo.paths$kpos1[i], limit = list(gene = 5, cpd = 2), bins = list(gene = 5, cpd = 2), na.col = "gray", discrete = list(gene = T, cpd = T)) pv.out <- pathview(gene.data = sel.genes, cpd.data = sim.cpd.data, pathway.id = demo.paths$sel.paths[i], species = "hsa", out.suffix = "sel.genes.cpd", keys.align = "y", kegg.native = T, key.pos = demo.paths$kpos1[i], limit = list(gene = 5, cpd = 1), bins = list(gene = 5, cpd = 10), na.col = "gray", discrete = list(gene = T, cpd = F))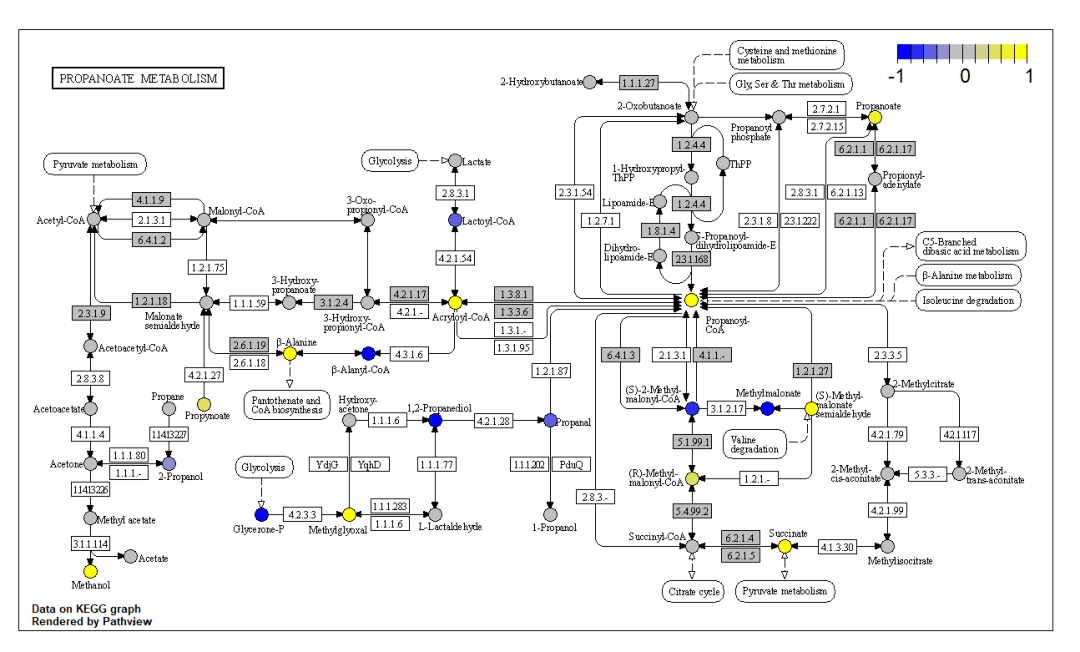 使用不同的ID格式 pv.out
<- pathview(gene.data = gene.ensprot, cpd.data = cpd.cas,
gene.idtype = gene.idtype.list[4], cpd.idtype = cpd.simtypes[2],
pathway.id = demo.paths$sel.paths[i], species = "hsa", same.layer = T,
out.suffix = "gene.ensprot.cpd.cas", keys.align = "y", kegg.native = T,
key.pos = demo.paths$kpos2[i], sign.pos = demo.paths$spos[i], limit =
list(gene = 3, cpd = 3), bins = list(gene = 6, cpd = 6))
使用不同的ID格式 pv.out
<- pathview(gene.data = gene.ensprot, cpd.data = cpd.cas,
gene.idtype = gene.idtype.list[4], cpd.idtype = cpd.simtypes[2],
pathway.id = demo.paths$sel.paths[i], species = "hsa", same.layer = T,
out.suffix = "gene.ensprot.cpd.cas", keys.align = "y", kegg.native = T,
key.pos = demo.paths$kpos2[i], sign.pos = demo.paths$spos[i], limit =
list(gene = 3, cpd = 3), bins = list(gene = 6, cpd = 6)) 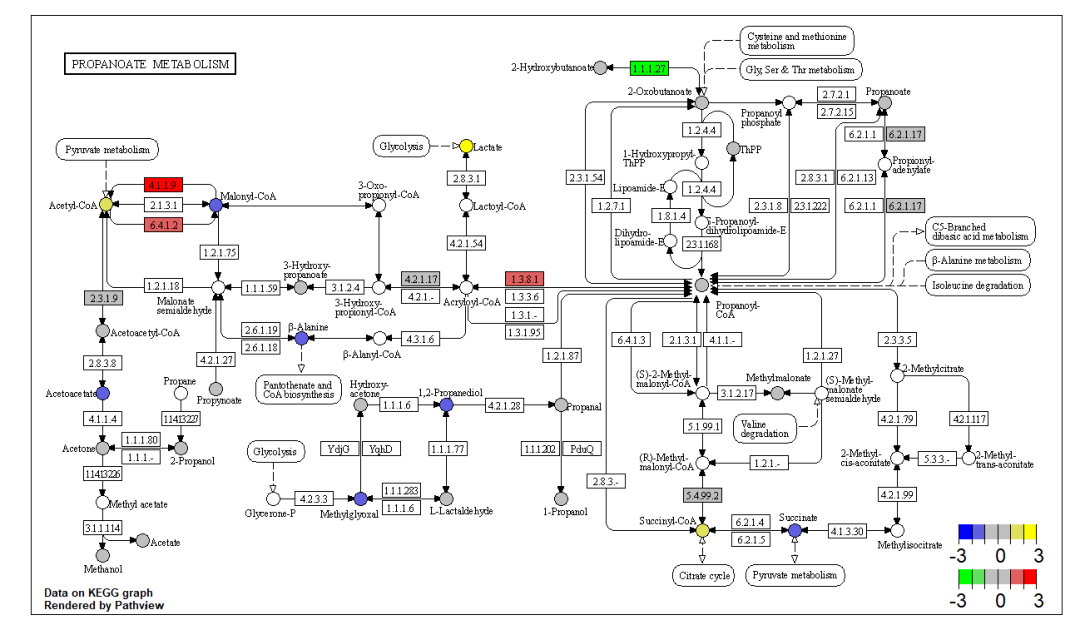 进行ID转换 id.map.cas
<- cpdidmap(in.ids = names(cpd.cas), in.type = cpd.simtypes[2],
out.type = "KEGG COMPOUND accession") cpd.kc <- mol.sum(mol.data =
cpd.cas, id.map = id.map.cas) id.map.ensprot <- id2eg(ids =
names(gene.ensprot), category = gene.idtype.list[4], org = "Hs")
gene.entrez <- mol.sum(mol.data = gene.ensprot, id.map =
id.map.ensprot) eco.dat.kegg <-
sim.mol.data(mol.type="gene",id.type="kegg",species="eco",nmol=3000)
eco.dat.entrez <-
sim.mol.data(mol.type="gene",id.type="entrez",species="eco",nmol=3000) 定义条件进行统计 data(korg)
head(korg) sum(korg[,"entrez.gnodes"]=="1",na.rm=T)
sum(korg[,"entrez.gnodes"]=="0",na.rm=T)
na.idx=is.na(korg[,"ncbi.geneid"]) sum(na.idx)
进行ID转换 id.map.cas
<- cpdidmap(in.ids = names(cpd.cas), in.type = cpd.simtypes[2],
out.type = "KEGG COMPOUND accession") cpd.kc <- mol.sum(mol.data =
cpd.cas, id.map = id.map.cas) id.map.ensprot <- id2eg(ids =
names(gene.ensprot), category = gene.idtype.list[4], org = "Hs")
gene.entrez <- mol.sum(mol.data = gene.ensprot, id.map =
id.map.ensprot) eco.dat.kegg <-
sim.mol.data(mol.type="gene",id.type="kegg",species="eco",nmol=3000)
eco.dat.entrez <-
sim.mol.data(mol.type="gene",id.type="entrez",species="eco",nmol=3000) 定义条件进行统计 data(korg)
head(korg) sum(korg[,"entrez.gnodes"]=="1",na.rm=T)
sum(korg[,"entrez.gnodes"]=="0",na.rm=T)
na.idx=is.na(korg[,"ncbi.geneid"]) sum(na.idx)
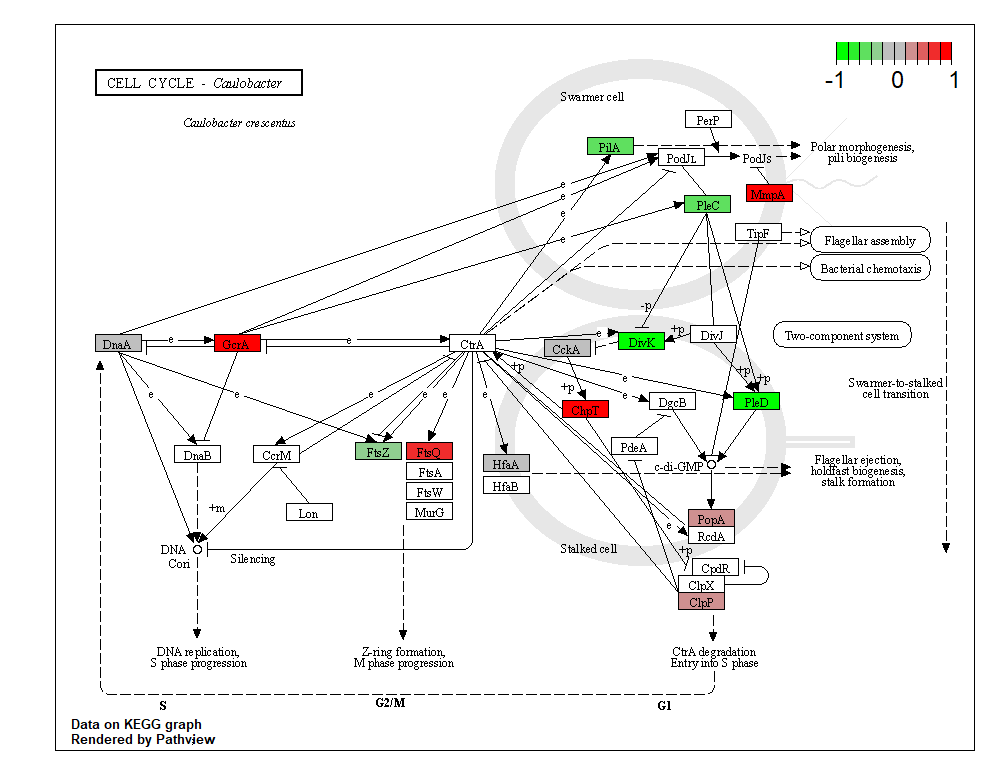
#画出人工绘制的通路图ko.data=sim.mol.data(mol.type="gene.ko", nmol=5000)
pv.out <- pathview(gene.data = ko.data, pathway.id = "04112",species = "ko", out.suffix = "ko.data", kegg.native = T)
 好了,以上就是使用Pathview画出高大上的基因与代谢通路热图的操作,希望对你有所帮助。如果对Pathview的使用有什么问题可以评论区留言,更多你不知道的生物学小工具教程及下载欢迎继续关注~
好了,以上就是使用Pathview画出高大上的基因与代谢通路热图的操作,希望对你有所帮助。如果对Pathview的使用有什么问题可以评论区留言,更多你不知道的生物学小工具教程及下载欢迎继续关注~
转自:唯誉智合
- 本文固定链接: https://maimengkong.com/image/1065.html
- 转载请注明: : 萌小白 2022年6月29日 于 卖萌控的博客 发表
- 百度已收录
