宏基因组学研究的对象是特定环境中的总DNA,不是某特定的微生物或其细胞中的总DNA,不需要对微生物进行分离培养和纯化,这对我们认识和利用未培养微生物提供了一条新的途径。
宏基因组测序无需PCR扩增,其实验流程见下图:
宏基因组测序无需PCR扩增,其实验流程见下图:
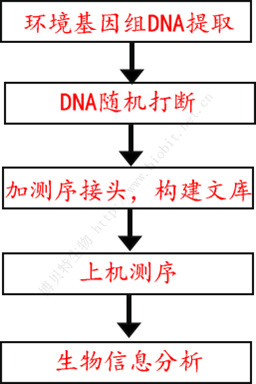
图1 宏基因组/宏转录组实验流程
宏基因组数据的标准分析流程通常包括质量控制、rRNA识别、序列组装 (assembly)、基因预测 (gene calling)、基因注释
(功能和生物来源的annotation)
等步骤(如下图)。其个性化分析多种多样,例如多样性分析(PCoA、NMDS等排序分析、PerMANOVA/Anosim分析)、LEfSE分析、与环境因子的关联分析
(Mantel test、方差分解分析VPA等)、网络分析,等等。
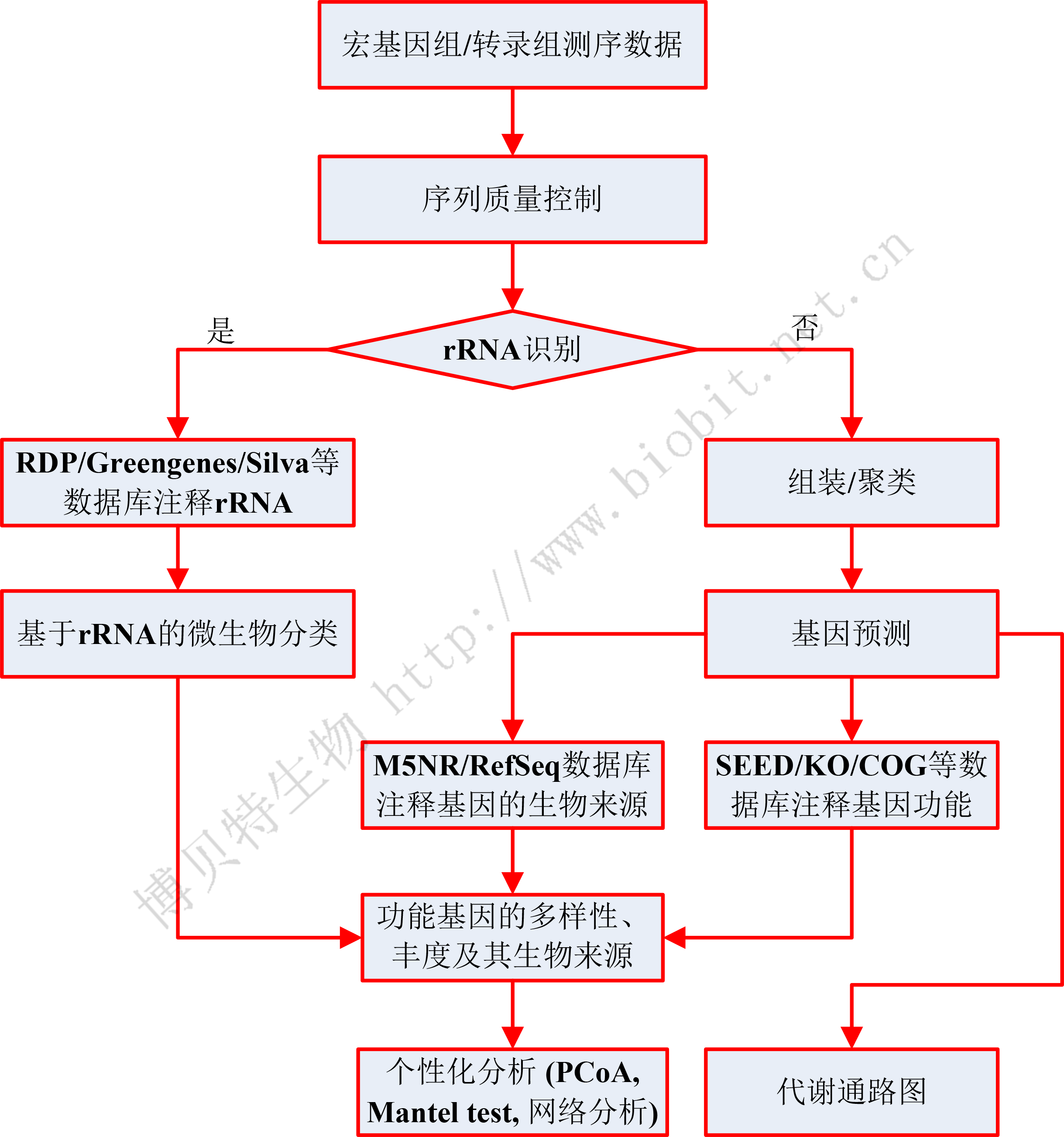
图2 宏基因组/宏转录组生物信息分析流程
宏基因组生物信息分析结果示例如下:
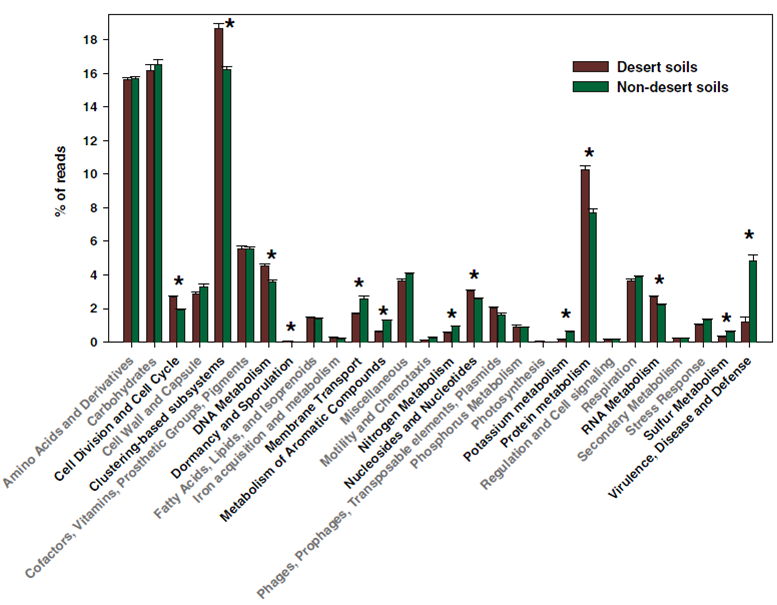
图3 用柱状图呈现不同处理之间功能类别(SEED subsystems功能库Level 1 水平)的丰度差异(Fierer et al. PNAS 2012)
图3 用柱状图呈现不同处理之间功能类别(SEED subsystems功能库Level 1 水平)的丰度差异(Fierer et al. PNAS 2012)
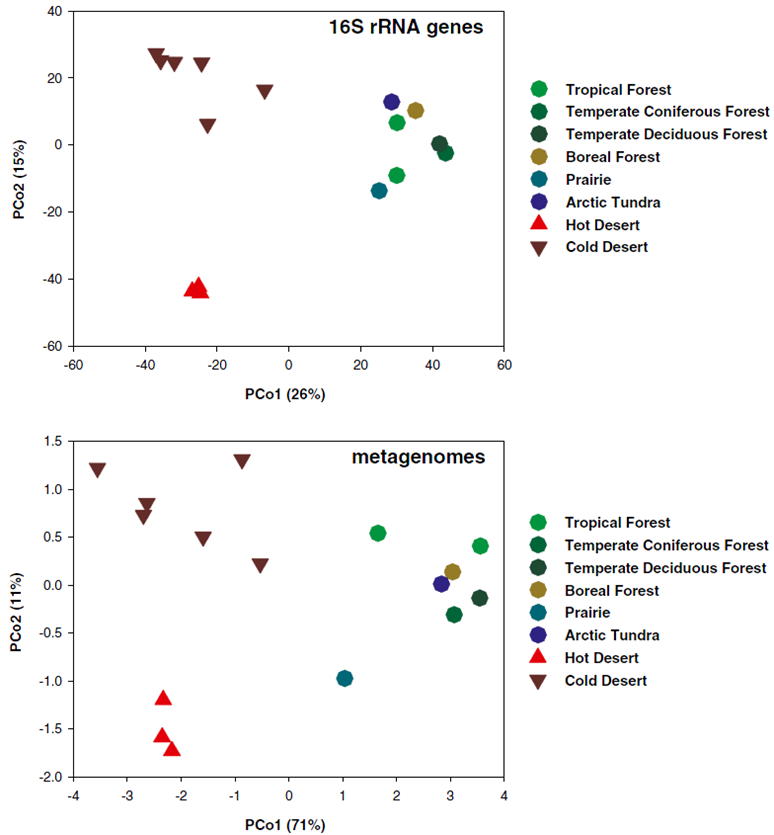
图4 用PCoA图呈现微生物群落(a. 16S rRNA)与功能(b. 宏基因组)在beta多样性上的差异(Fierer et al. PNAS 2012)
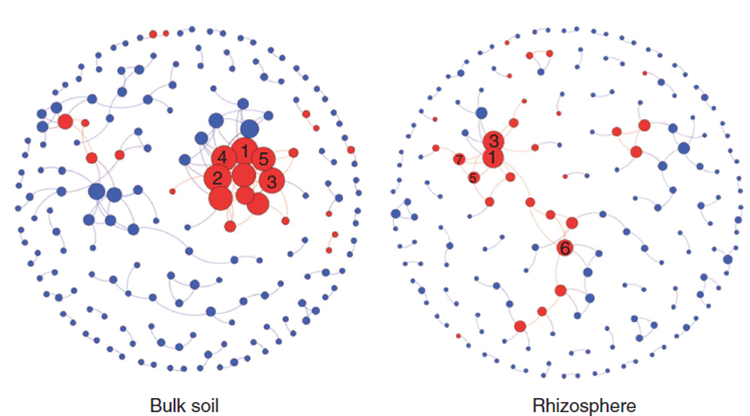
图5 比较不同处理(非根际土与根际土)之间的微生物功能网络(Menders et al. ISME J 2014)
图中蓝色节点表示功能,红色节点为细菌门,连线表示两节点之间存在显著相关。
图中蓝色节点表示功能,红色节点为细菌门,连线表示两节点之间存在显著相关。
- 本文固定链接: https://maimengkong.com/zu/862.html
- 转载请注明: : 萌小白 2022年4月3日 于 卖萌控的博客 发表
- 百度已收录
