今天给大家带来的复现比较经典文章是来自2区的Frontiers in Immunology杂志,影响因子高达7.5分!
题目是: Hepcidin Upregulationin Lung Cancer: A Potential Therapeutic Target Associated With Immune Infiltration

要素拆解
题目:肺癌中铁调素升高可以作为潜在的免疫相关的治疗靶标
疾病:肺癌(包含肺腺癌,肺鳞癌)
物种:人
数据来源:TCGA,GEO
背景知识
铁调素(hepcidin)是由肝脏合成并分泌的富含半胱氨酸的抗菌多肽,在免疫过程中能够大量表达。
参与免疫反应,在机体内铁平衡的调节中起到负性调节的作用,其在铁代谢疾病的相关临床应用中有一定疗效。
铁调素主要由肝细胞合成,并作为25个氨基酸的肽即铁调素-25被分泌到循环中,最后由肾脏清除。
在生理状态下,当机体内铁过载出现时,铁调素在肝脏中的合成及分泌会增加,从而抑制力肝脏中铁的释放及肠道铁的吸收,避免了铁过量产生过多活性氧致细胞损伤或死亡。
数据解读
本文一共有8张图,2张表,7张附图,数据量非常扎实
Figure 1:分析铁调素在肺癌中的mRNA表达
Figure 2:分析铁调素在肺癌中的蛋白表达
Figure 3:肺癌病人铁调素表达与临床相关性
Figure 4:铁调素预后相关分析
Figure 5:鉴定与铁调素互作的基因
Figure 6:GO/KEGG通路富集分析
Figure 7:免疫浸润相关分析
Figure 8:基于免疫细胞的铁调素表达是否和预后相关的分析
Table 1:利用TIMER数据库对铁调素和基因标志物的相关性分析
Table 2:利用TIMER数据库对铁调素和不同T细胞的gene marker的相关性进行分析
复现工具
仙桃学术工具:https://www.xiantao.love/products
UALCAN数据库:http://ualcan.path.uab.edu/
GEPIA数据库:http://gepia.cancer-pku.cn/index.html
cBioportal数据库:https://www.cbioportal.org/
TIMER数据库:http://timer.cistrome.org/
CIBERSORT算法数据库:https://cibersort.stanford.edu/
Kaplan-Meier Plotter数据库:http://kmplot.com/analysis/index.php?p=background
PrognoScan Database数据库:http://dna00.bio.kyutech.ac.jp/PrognoScan/index.html
GeneMANIA数据库:http://genemania.org/
HPA数据库:https://www.proteinatlas.org/
1: 研究的靶基因经典,在pubmed上以关键词hepcidin搜索,已经有542篇文章,说明这个靶基因不是特别新的蛋白,依然是可以发生信套路文章的。
如果再聚焦到本文研究的癌种,肺癌,依然已经有16篇文章,由此可见,利用生信的方法进行分析,即便靶基因不是特别具有创新性,也是可以发文章的,甚至分数还不低哦
2: 生信分析经典,文章里的方法,首先看靶基因的表达,临床相关性,生存等都是非常经典的分析方法,随便代入哪个到我们自己的分析都是很适用的,分分钟复现出一篇属于自己的SCI。
文章复现
Figure 1:在肺癌中检测到铁调素(Hepcidin)高表达
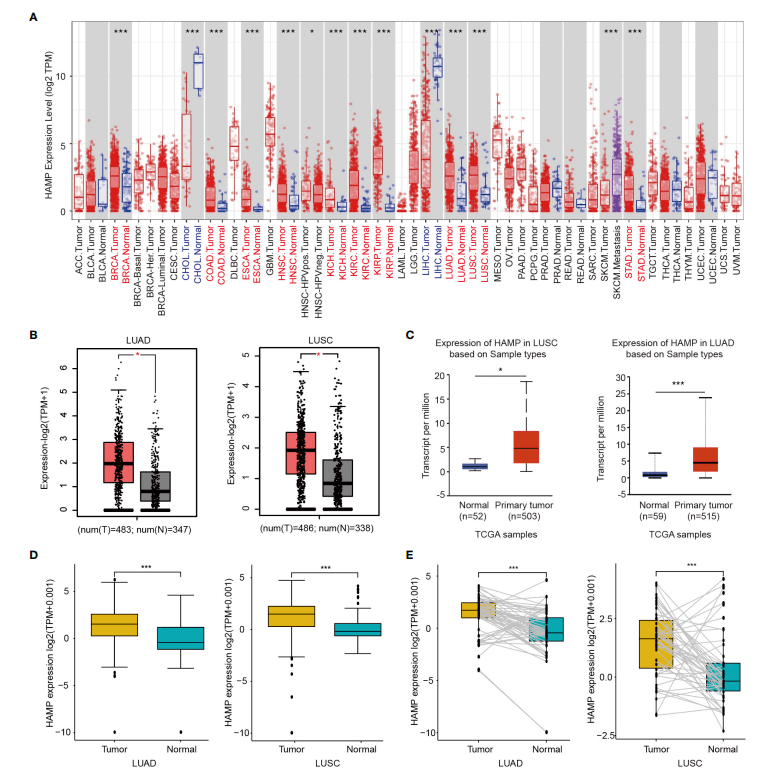
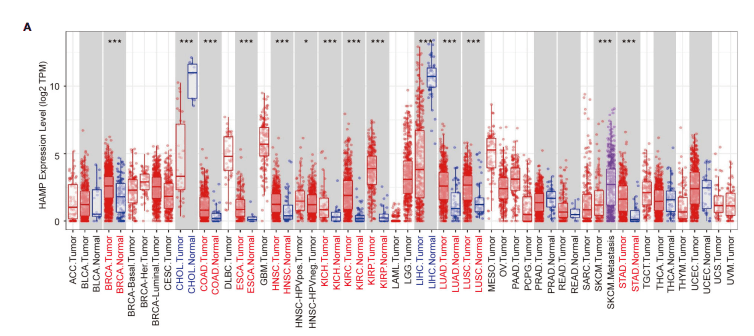
Figure 1A:利用TIMER数据库分析了Hepcidin在泛癌中的表达
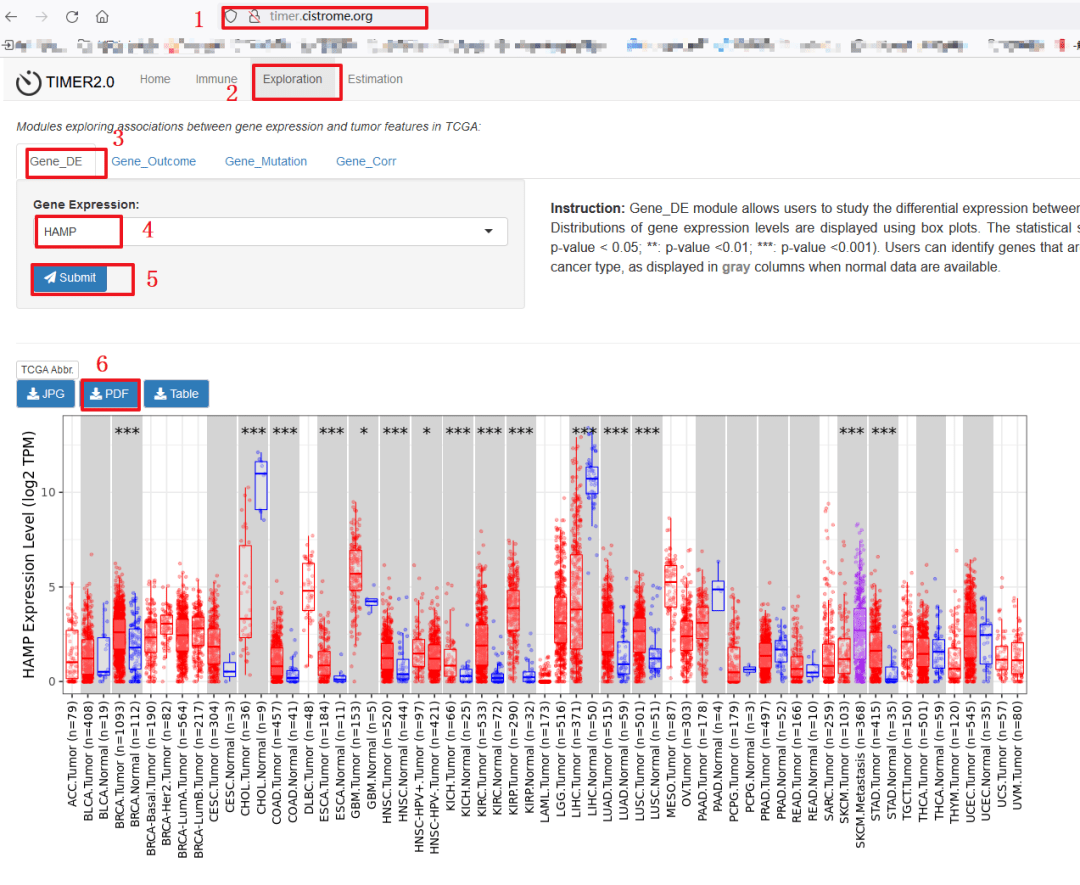
复现具体步骤如下:
1:点击TIMER数据库网址,http://timer.cistrome.org/
2:选择【Exploration】模块
3:接着选择【Gene_DE】模块
4:输入我们的目标基因【HAMP】
5:点击【Submit】
6:网速好的情况下,即刻可以出图,我们直接点击下载PDF备用,即可得到和原文一模一样的Figure 1a
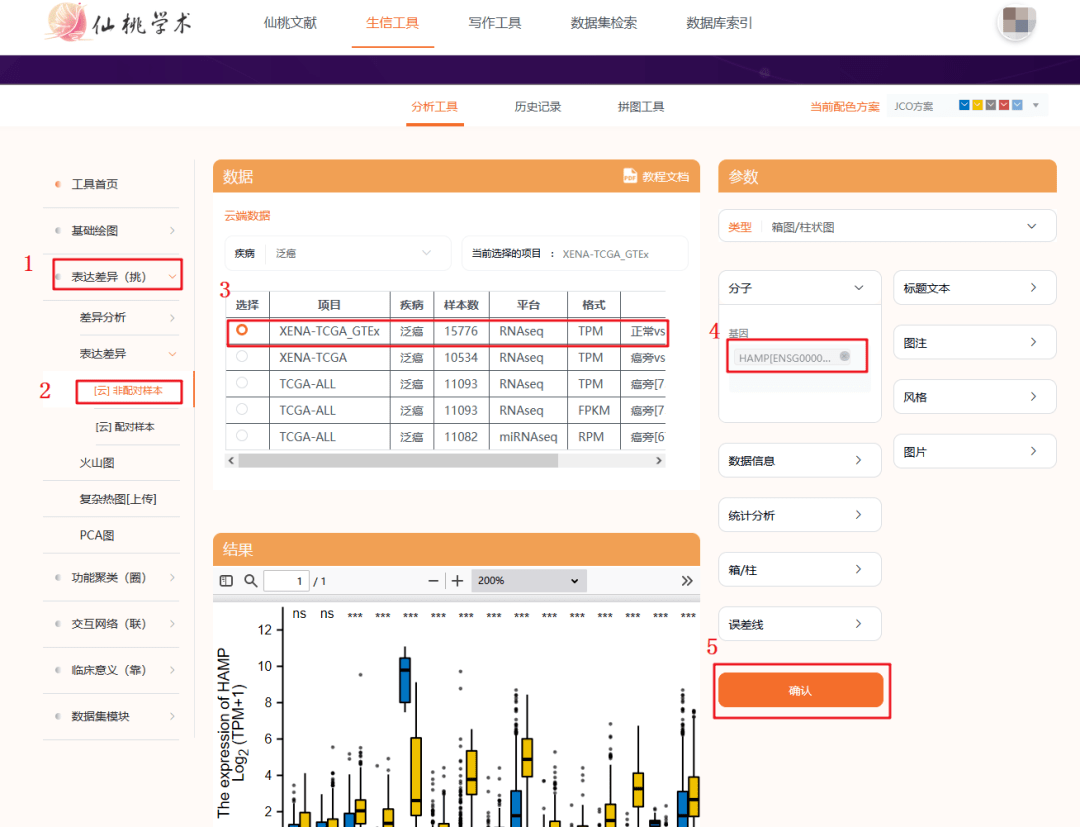
第二种研究靶基因在泛癌中的表达方法:利用仙桃工具进行复现,具体方法如下:
1:进入仙桃工具,选择【表达差异】模块
2:选择【非配对样本】
3:选择泛癌【XENA-TCGA-GTEx】
4:输入靶基因【HAMP】
5:点击【确认】,即可得到靶基因在泛癌中的表达,出的图形非常好看,美观。
接着,我们来复现Figure 1B:利用GEPIA分析靶基因表达
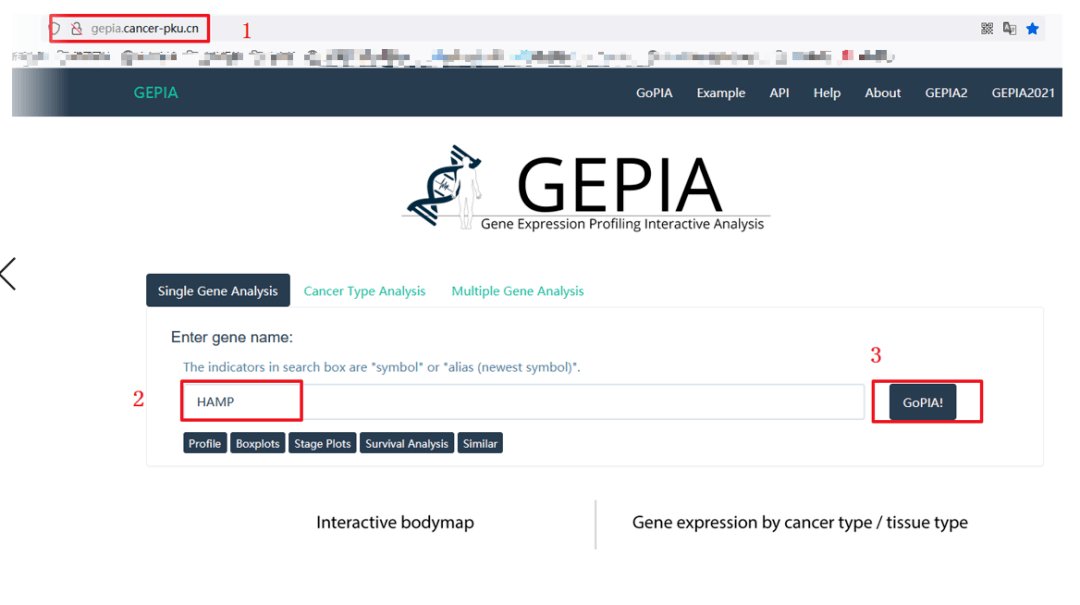
1:点击GEPIA网址,http://gepia.cancer-pku.cn/
2:输入靶基因【HAMP】
3:点击【GoPIA】
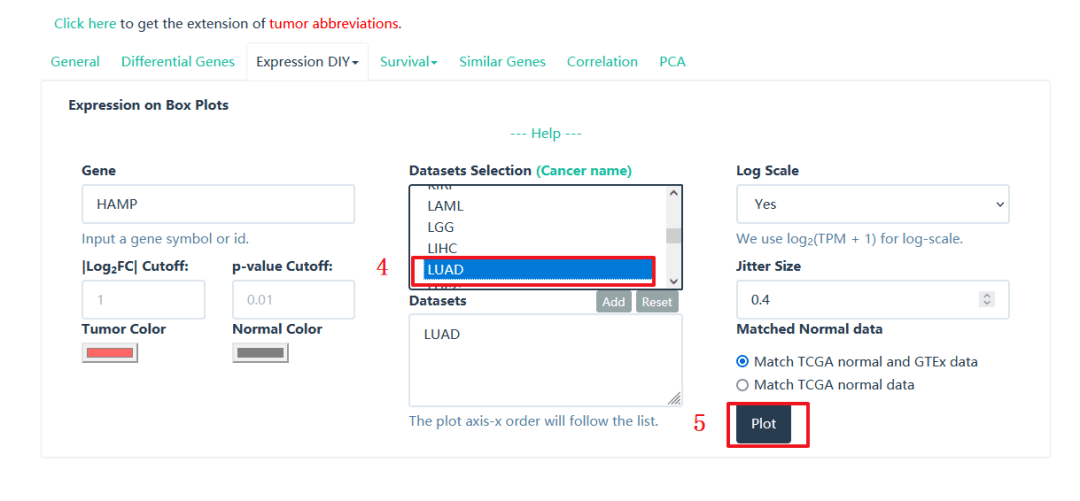
4:选择研究的癌种【LUAD】
5:点击【Plot】,即可出图
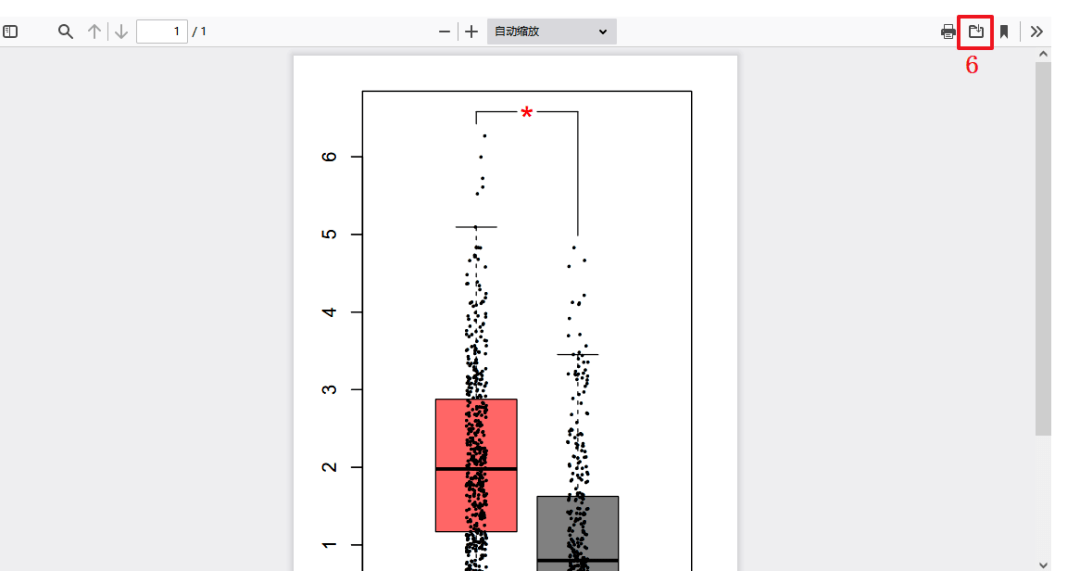
6:点击下载按钮,即可得到和Figure 1B LUAD一模一样的图,同样的,LUSC只需在第4步,将癌种修改为LUSC即可完整得到Figure 1B。
接着我们来复现Figure 1C:作者利用UALCAN数据库,分析HAMP表达
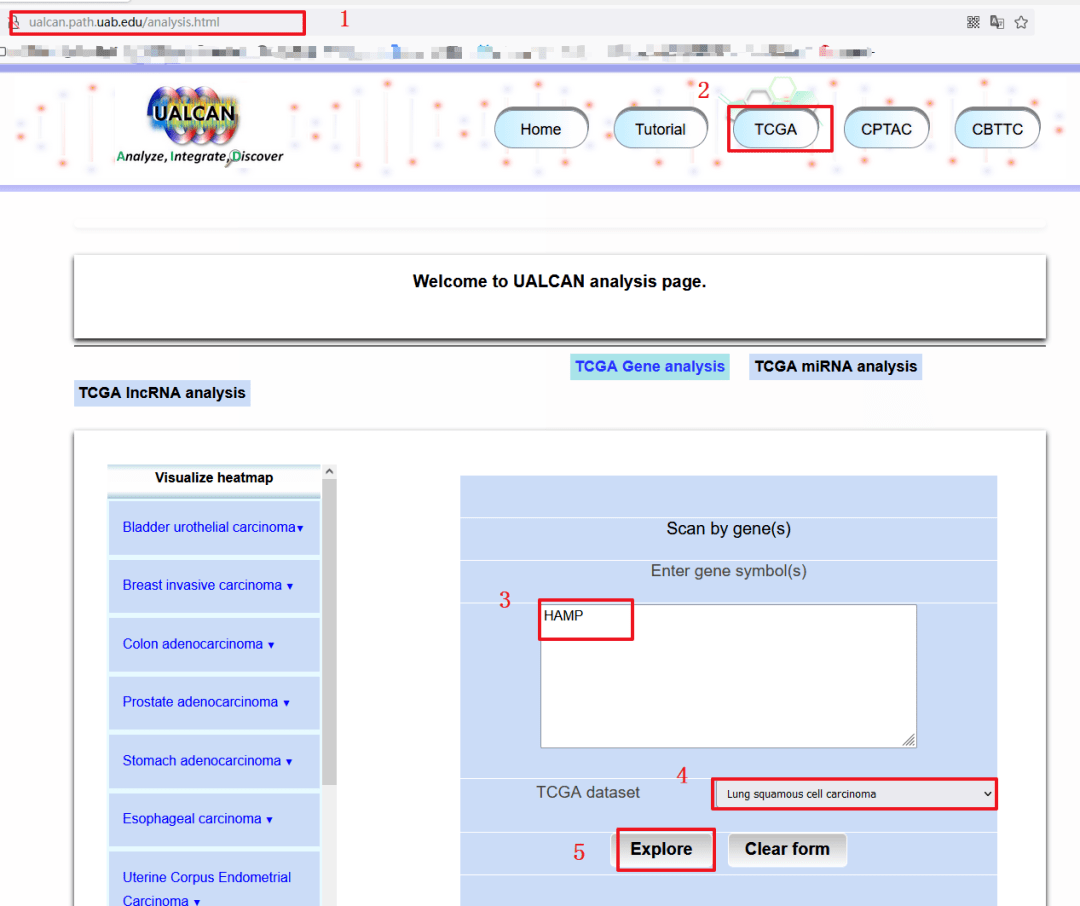
1:首先输入网址:http://ualcan.path.uab.edu/
2:选择【TCGA】模块
3:输入研究的靶基因【HAMP】
4:选择我们需要研究的癌种【LUSC】
5:点击【Explore】
6:作者直接将下方的显著性数值转换成星号*放在图中,这步操作我们可以在AI里完成
7:如需下载,我们点击这个框里下载按钮即可,LUAD操作和LUSC基本一致,只需在第4步将【LUSC】换成【LUAD】即可。这样Figure 1C复现完毕。
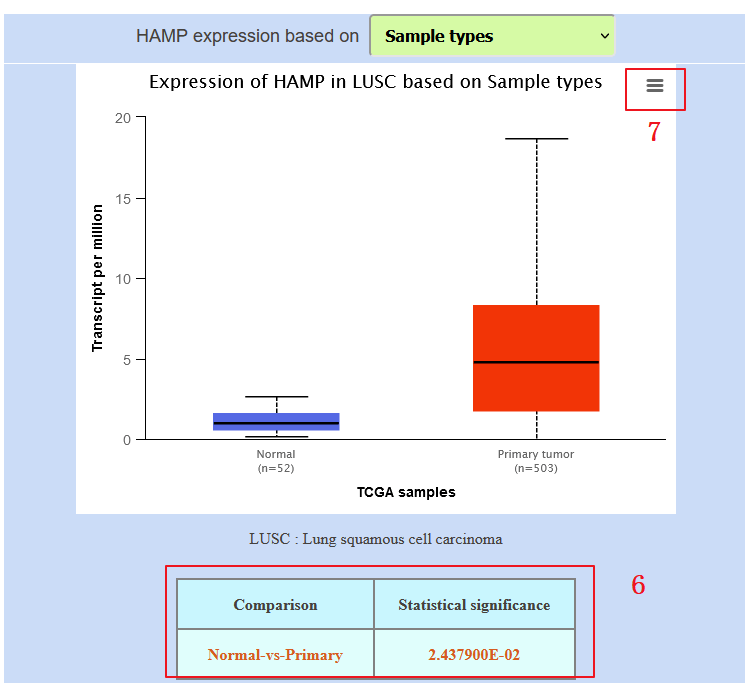
现在我们来复现Figure 1D:第4种方法分析靶基因表达
1:打开仙桃学术网址
2:点击【生信工具】
3:【高级版】--【立即使用】
(注:免费版和基础版都可以进行统计和可视化,由于高级版功能最全,这里选择高级版作为演示)
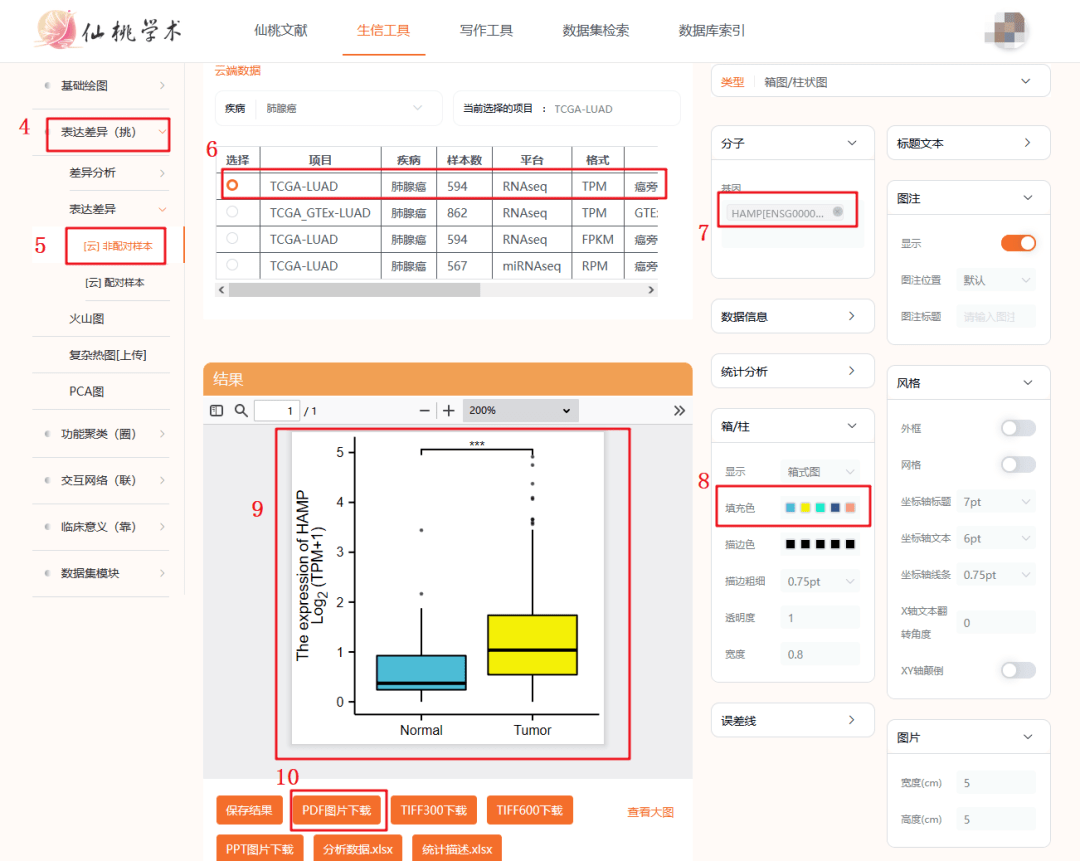
4:选择【表达差异】模块,
5:Figure 1D展示的是非配对样本,所以我们在这一步选择【非配对样本】
6:选择癌种【LUAD】,同样的方法稍后选择【LUSC】
7:输入我们的目标分子【HAMP】
8:关于填充色,我们工具默认的是蓝红,文章中癌和癌旁分别展示的黄,蓝,所以我么在配色这里修改一下即可
9:点击确认之后,图展示在此框框区域内
10:我们可以点击图片下载备用,LUAD即完整复现,LUSC复现步骤非常类似,在第6步修改癌种即可。

接下来我们来复现Figure 1E:配对样本的靶基因表达
其实和Figure 1D非常类似,前一张图展示的是Figure 1D非配对样本的HAMP表达量,后一张图展示的是癌和癌旁配对的HAMP表达量,具体复现步骤如下:
1:选择仙桃工具的【配对样本】
2:选择癌种【LUAD】
3:输入目标基因【HAMP】
4:点击确认
5:图形即可出现在中间区域,点击PDF格式下载待用,即可得到Figure 1E,LUSC同样的方法,记得选择配对样本即可,拼接起来即可得到Figure 1E
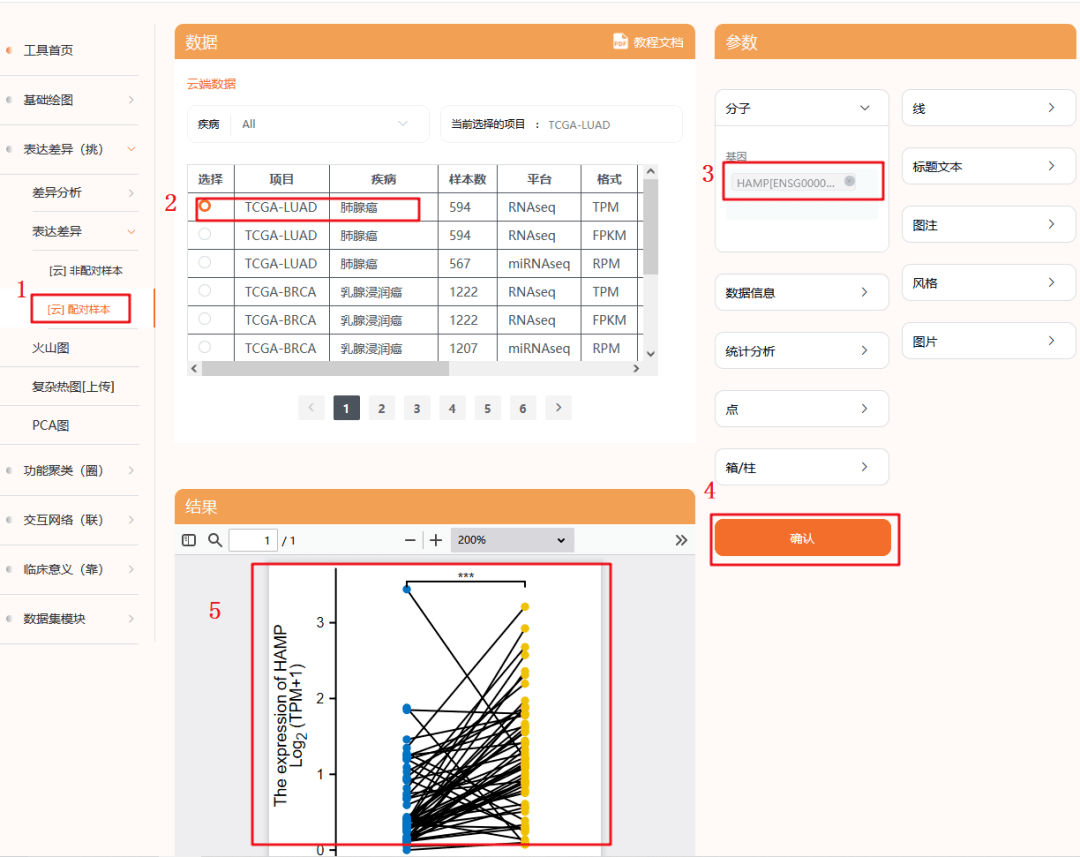
Figure 2:铁调素在肺癌病人的蛋白表达
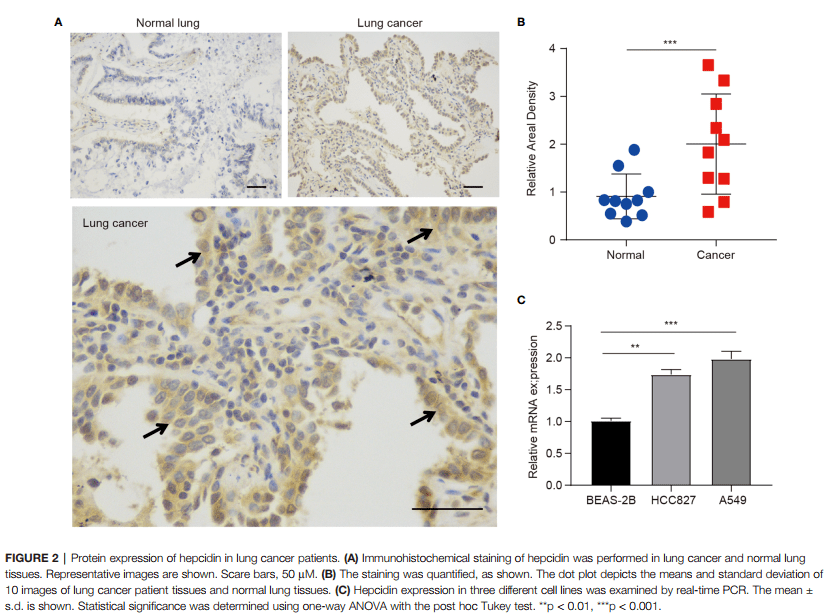
这部分作者是自己用自己收集的样本做的免疫组化,如果小伙伴没有抗体,或者收集不到临床组织,我们可以不花钱也得到免疫组化的结果哦!
我们利用human protein atlas这个数据库,网址如下:https://www.proteinatlas.org/search/HAMP
由于HPA网站没有收录hepcidin这个蛋白,所以我们不能在线获取了,但是小伙伴手里的目标基因应该大部分都是收录在内的,大家可以试一试,省钱又省精力~
Figure 3:利用UALCAN数据库分析铁调素表达和临床因素之间的相关性

关于这个数据库小编很想好好介绍一下,这个数据库简直就是0基础发文章的神器,他是一个全面的,用户友好的,交互式的癌症组学数据分析网络资源
为访问公开癌症组学数据库(TCGA, MET500, CPTAC)提供了极大的便利,不用下载任何数据便能轻松分析并导出结果图片。
UALCAN主要用途有一下几个方面:
1:可以快速分析出目标基因的mRNA表达情况
2:获取protein-coding,miRNA-coding,lincRNA-coding genes表达与患者生存信息的分析结果
3:分析基因的启动子甲基化状态
4:通过差异基因热图筛选感兴趣基因
5:对pan-cancer基因进行表达分析等
这个数据库出图快,包含信息多,是一个非常棒的0基础发文利器。
关于Figure 3具体复现步骤如下:
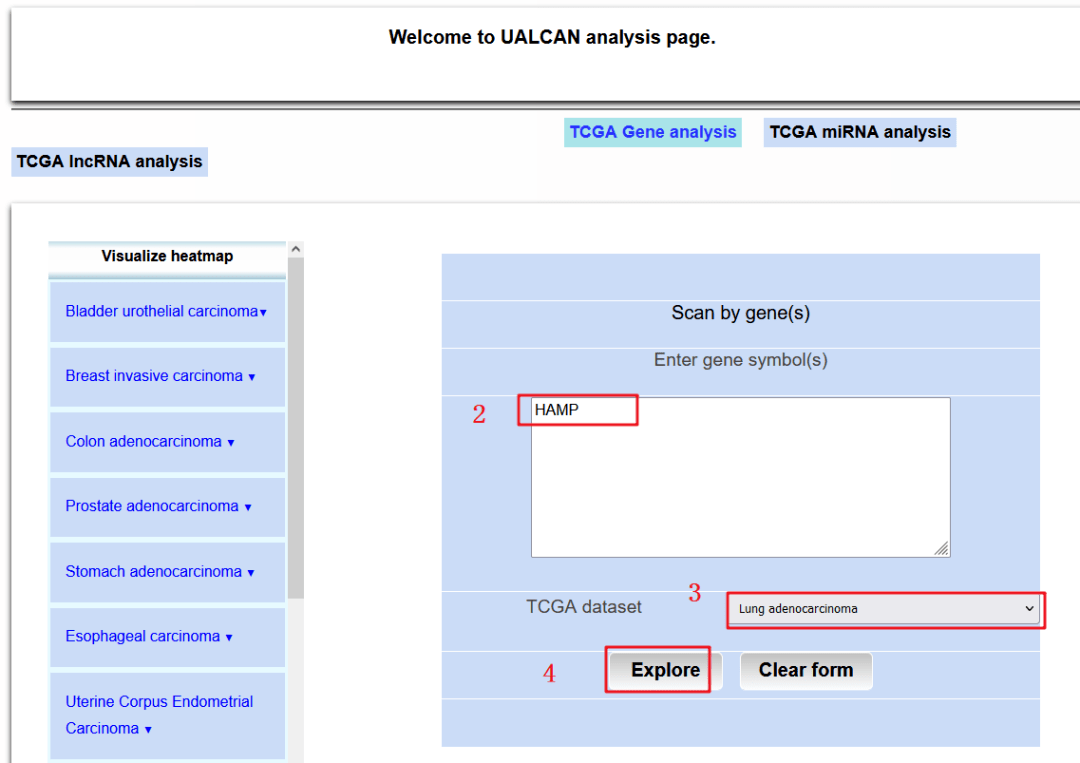
1:打开UALCAN数据库,http://ualcan.path.uab.edu/
2:输入我们的目标基因【HAMP】
3:输入我们研究的癌种【LUAD】
4:点击【Explore】
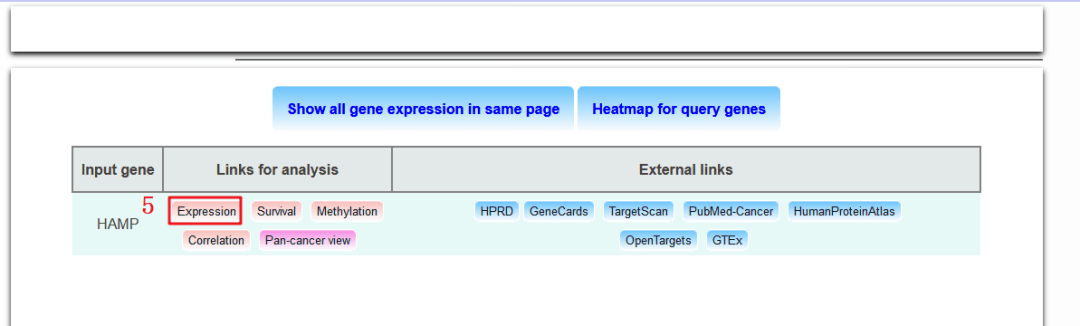
5:点击【Expression】,分析其表达
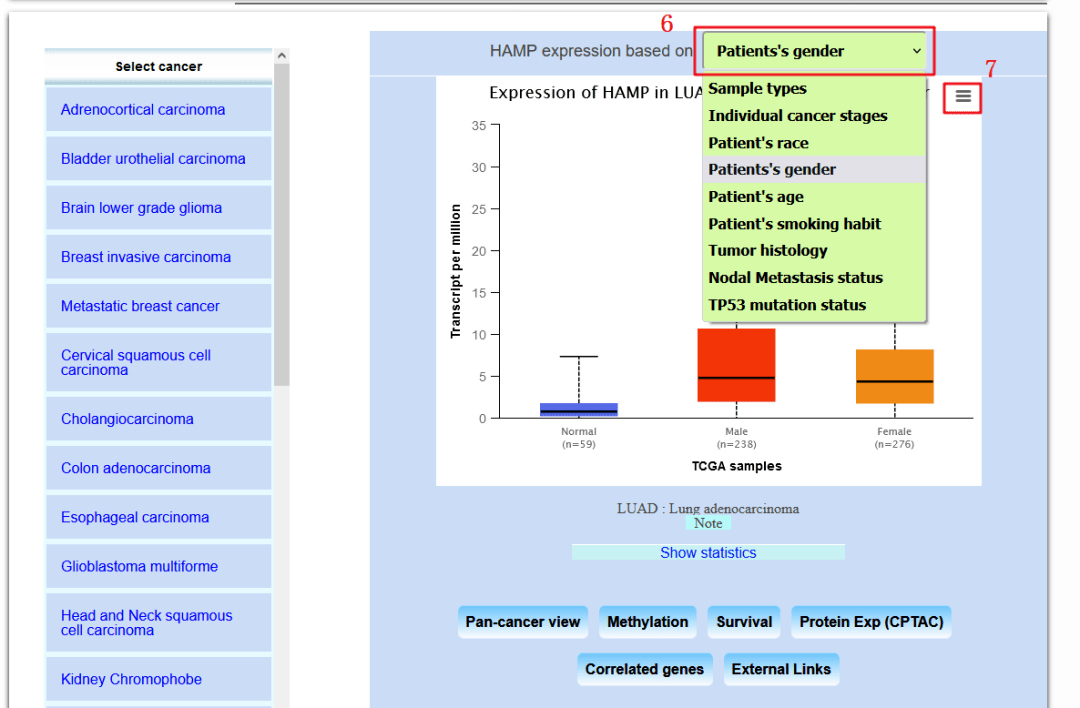
6:我们复现Figure 3A为例,基于LUAD肺癌病人性别,HAMP的表达情况。
7:这一步点击这个按钮可以下载本图,这样Figure 3A上面这张图就完整复现出来,下面这张图是关于LUSC病人,基于病人性别HAMP的表达情况,只需在第3步中,将LUAD换成LUSC即可。
接着复现Figure 3B,3C,同样的操作流程,在第6步中,将【Patient’s gender】分别换成【[individual cancer stages]和【metastasis status】即可,大家可以自己动手尝试一下。
Figure 3这张图我们仙桃学术也是完全可以复现的哦,更加方面快捷,让我们一起来看看。
1:点击仙桃学术,选择【临床意义(靠)】
2:选择【临床相关性模块】
3:选择癌种【TCGA-LUAD】
4:输入我们的目标分子【HAMP】
5:临床分组,我们首先复现Figure 3A,也就是病人性别对靶基因表达的影响,根据Normal,Male,Female分组。
6:这里可以自定义一些统计学方法进行分析,一般默认是Kruskal-Mallis test,这种方法又叫克鲁斯卡沃力斯测试,非参数检验方法。
检验是利用多个样本的秩和来推断各样本分别代表的总体的位置有无差别,一般多用于多组样本不满足正态性的情况。
其他一些统计学方法罗列在下,可以参考一下(具体可以参考仙桃的教程文档哦)
7:点击确认后,即可出图,我们可以看到分析出来的图形,在【male】和【female】之间没有显著相关性,放在图中不太好看,这时候仙桃的优越性就体现出来了
我们可以个性化将这条线去掉,返回头来看一下第6步,我们可以将【分组对比】中第三个去掉
点击X以后,我们重新点击确认,即可出现和原文一模一样的图,颜色还可以更改成符合自己审美的颜色,比UALCAN选择性更多,忍不住感叹一句,仙桃真香呀~
同样的步骤,将【LUAD】换成【LUSC】,【patient’s age】分别换成【individual cancer stages】和【nodal metastasis status】即可完整得到Figure 3。
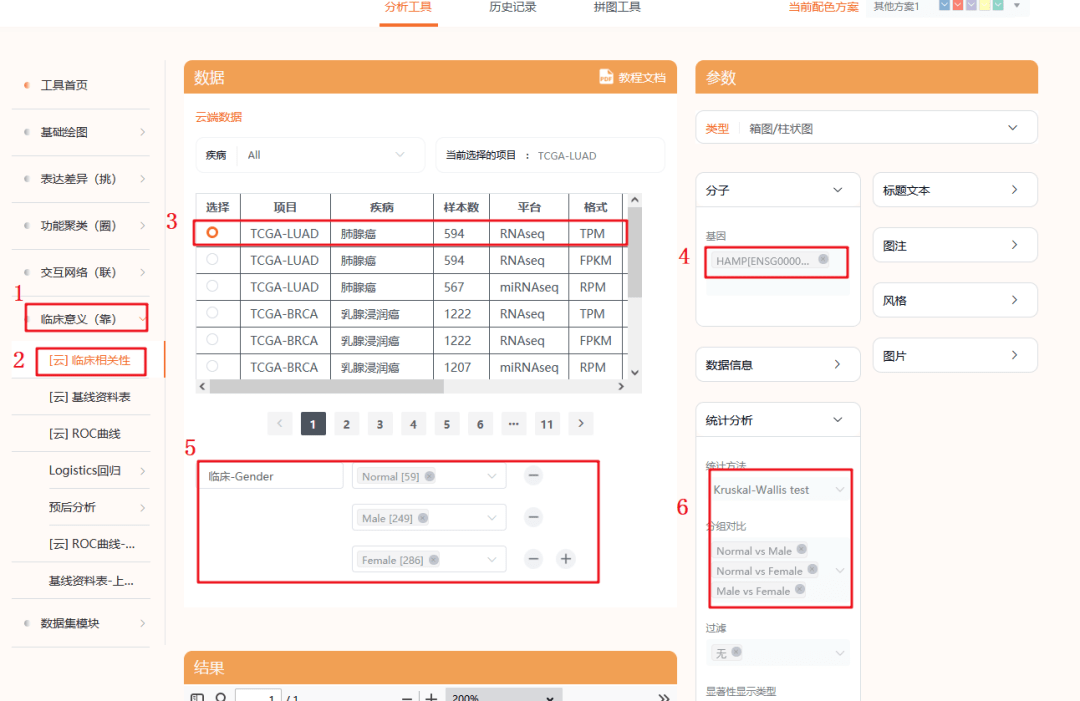

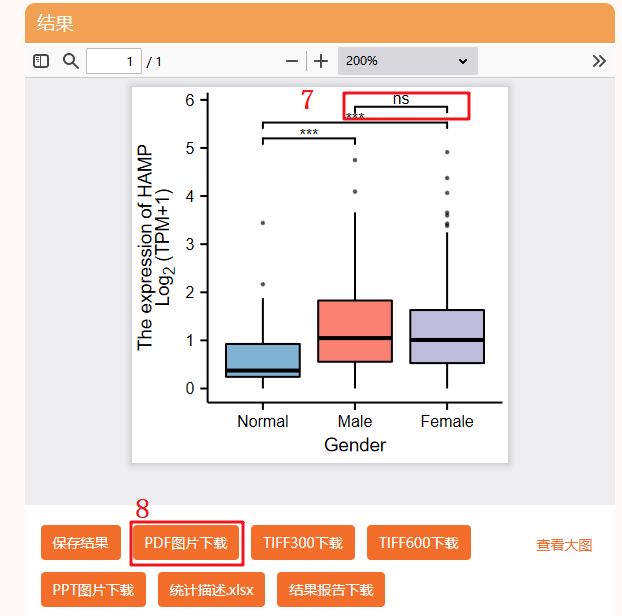
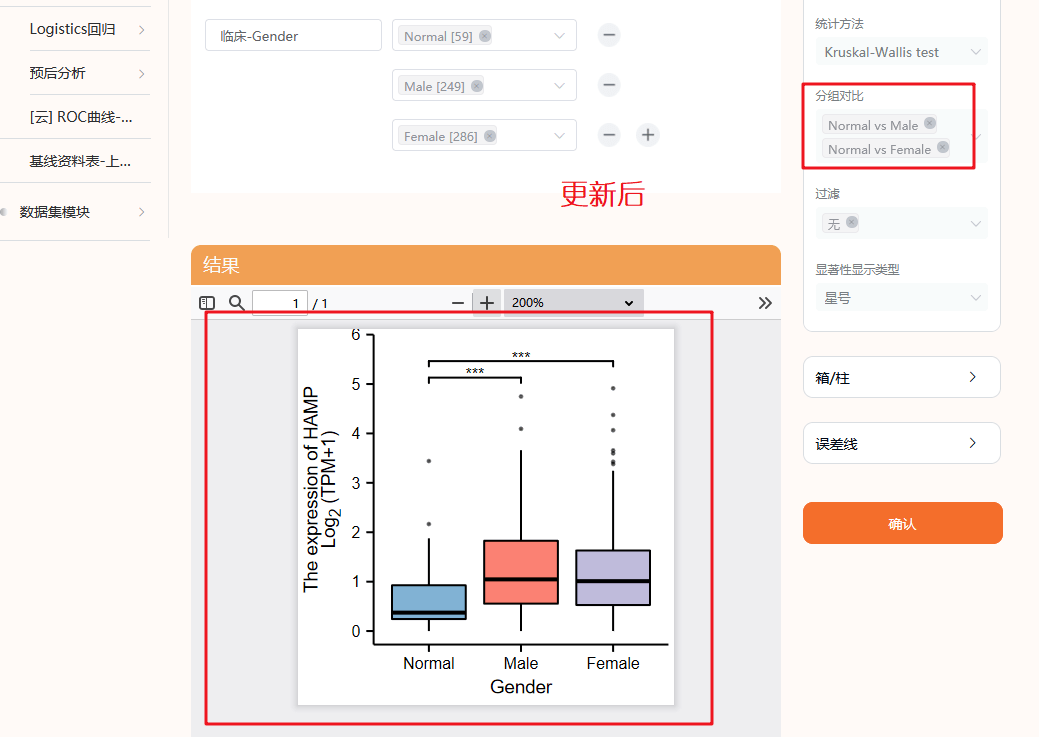
Figure 4,作者分析了靶基因hepcidin的生存预后
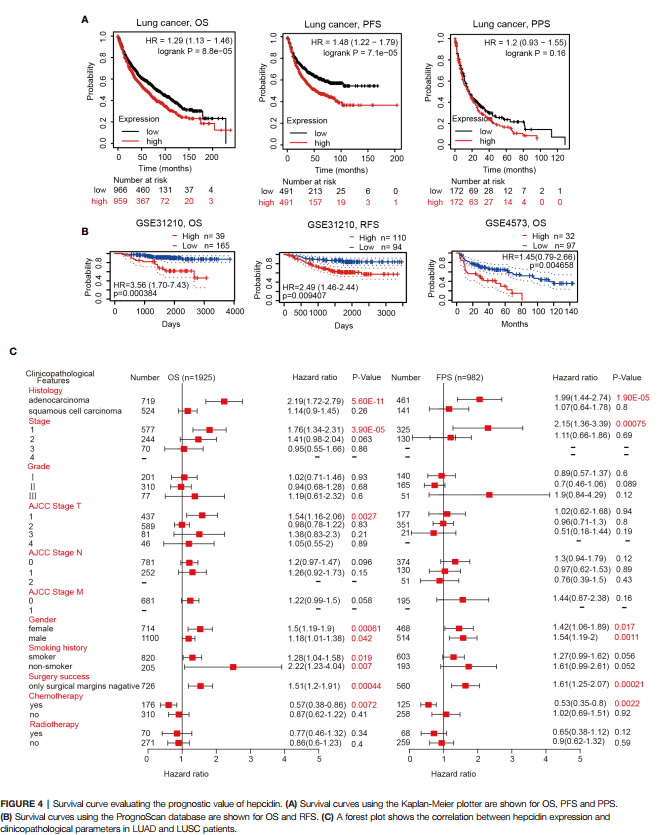
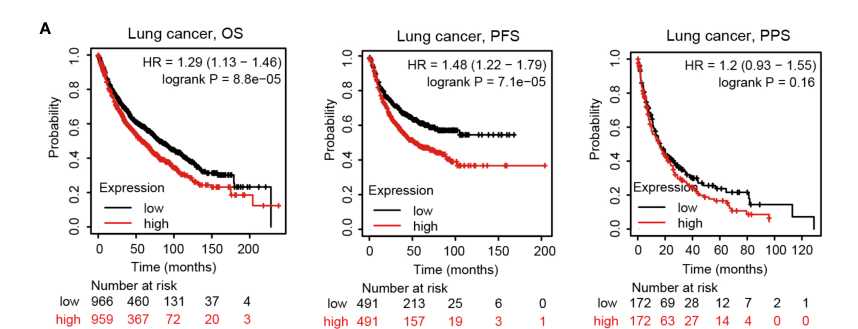
首先我们来复现Figure 4A,本张图是由KM-Plotter完成,作者分别分析靶基因在肺癌当中的OS,PFS,PPS不同生存状态下,HAMP高低表达对生存的影响。
具体复现步骤如下:
1:打开KM-plotter数据库网址,
http://kmplot.com/analysis/index.php?p=service&cancer=lung,输入目标基因【HAMP】
2:先选择【OS】,随后选择【PFS】,【PPS】
3:点击【Draw Kaplan-Meier plot】
4:红框框出来的P值和原文比对后一模一样,其实文章其实可以更加严谨一点就是在生存分析这一块,依然分组,将肺癌分成【LUAD】和【LUSC】,因为前面3张图,无论是表达,还是临床相关性分析都是将肺癌分为【LUAD】和【LUSC】,生存分析这里反而不分组了,就很突兀。
5:点击下载PDF格式文件,即可得到Figure 4A的第一张图。
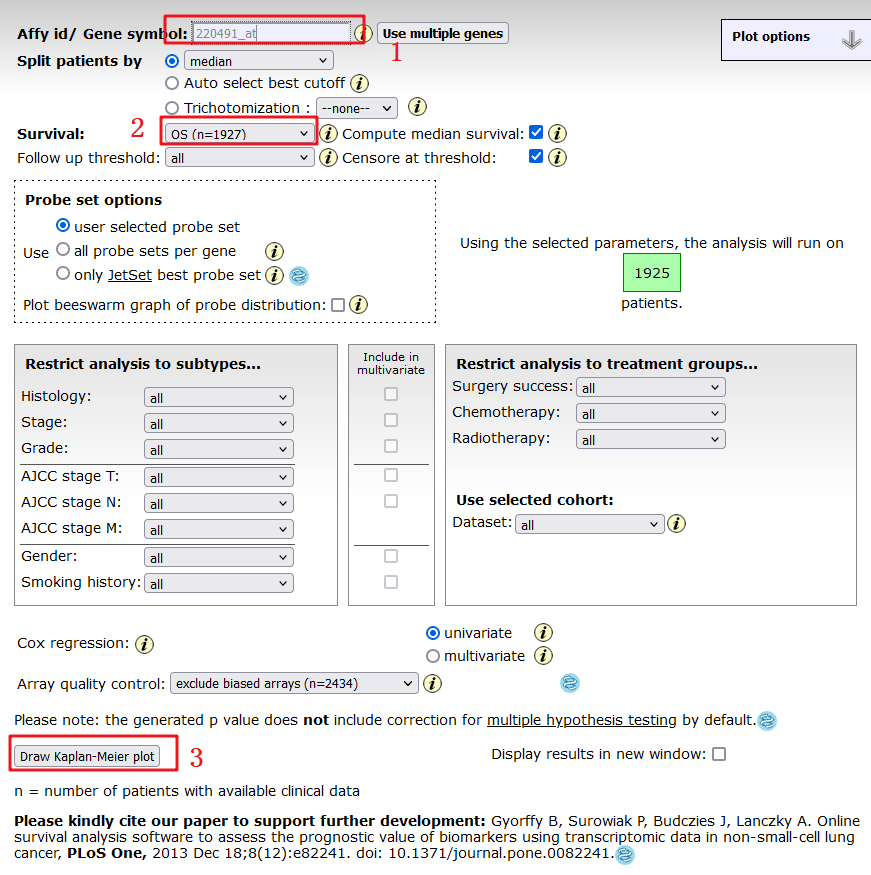
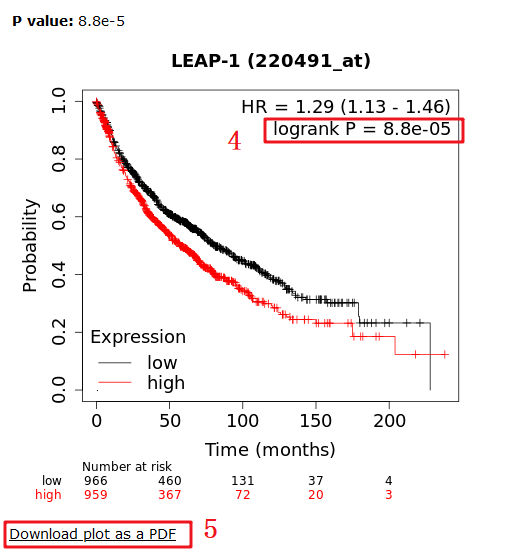
Figure 4A后面两张图还是同样的步骤,在第2步将【OS】,分别换成【PFS】,【PPS】即可,拼接起来即可得到Figure 4A.
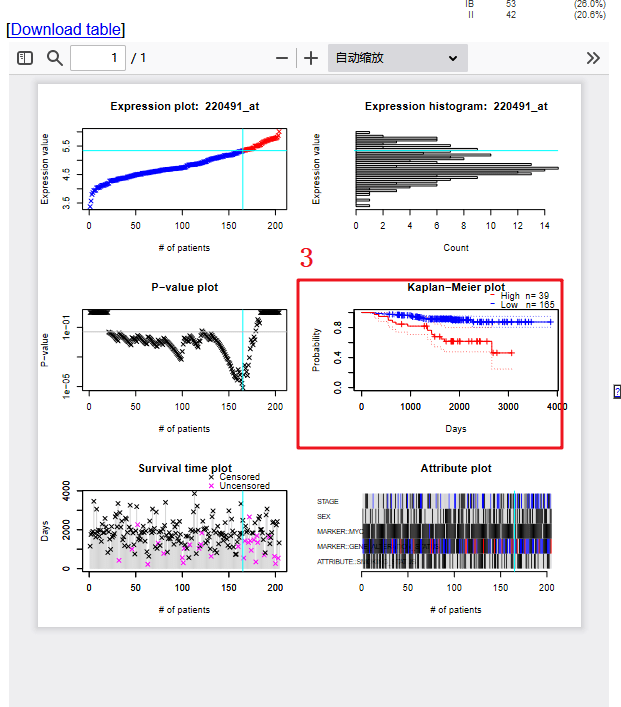
接着我们来复现Figure 4B,作者利用了prognoscan这个数据库
这个数据库是肿瘤生信文章分析基因预后的宠儿,它整合了大量带有预后信息的芯片数据集,站内基本上包括了大部分的肿瘤数据,可以用来分析基因表达与患者预后的关系,例如总体生存期(OS)和无病生存期(DFS)。
在PrognoScan创立前,已有许多带有临床注释信息的癌症芯片数据集向大众开放,但对各个数据库来源的数据集信息之间并不联通,还有各种限制难以访问。
为了充分利用公共资源,2009年日本人开发了 PrognoScan。
数据库通过大量收集公开的癌症芯片数据集来探索基因表达与患者临床预后(如总体生存期(OS)和无病生存期(DFS)等)之间的关系,提供了最佳的整合结果,给使用者提供了许多便利。
具体复现步骤如下:
1:进入prognoscan数据库,输入靶基因【HAMP】,点击【submit】
2:我们找到LUAD这个癌种,数据库中列举了几个GSE数据集,找到文章中提到的GSE31210,红框所示,点击进去。
3:出来的这一张图包含了很多信息,我们选择我们需要的KM生存曲线图即可,剩下2张小图也是同样的方法,拼接起来即可得到Figure 4B。

现在我们来复现Figure 4C,作者这张森林图是利用KM-plotter数据库得到。
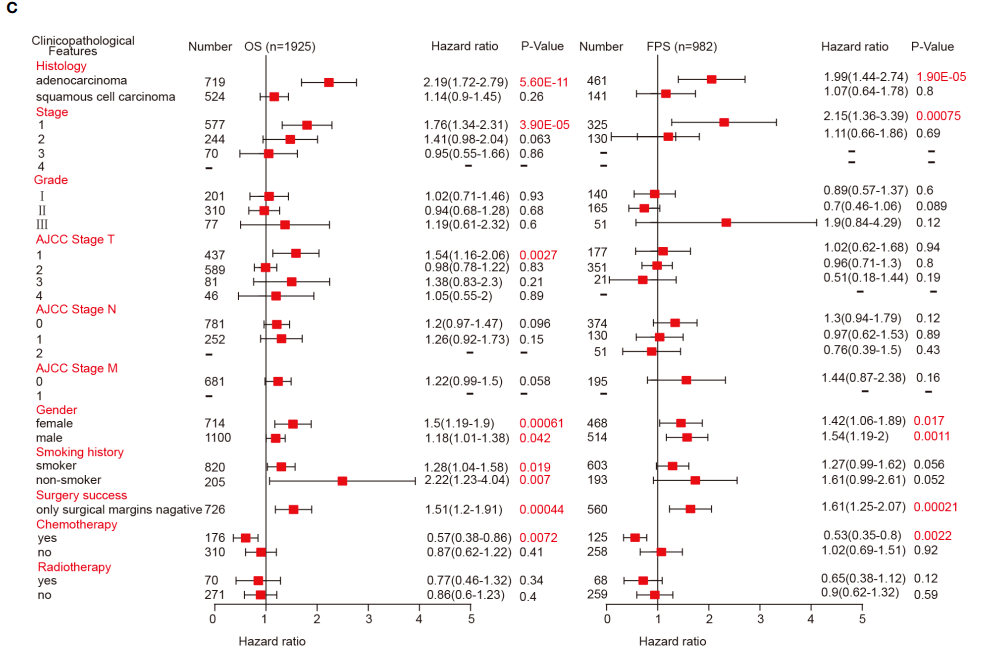
具体复现步骤如下
我们主要以左边为复现对象,首先是获取数据,通过观察可知,分为Histology,Stage,Grade, AJCC Stage T,AJCC stage N,AJCC stage M,Gender,Smoking history,Surgery success,Chemotherapy,Radiotherapy 11个部分。
我们首先获取Histology数据
1:进入KM-plotter数据库,输入靶基因【HAMP】
2:选择【LUAD】
3:点击【Draw Kaplan-Meier plot】
4:出来的图中包含的信息有 HR=2.19(1.72-2.79),logrank P=5.6e-11,将这些信息摘录下来即可。
5:我们做这森林图可以不下载,看个人需要
6:我们继续将Histology数据提取完,我们接下来选择【LUSC】这个选项
7:在这一步中可以得到包含524个样本
8:点击【Draw Kaplan-Meier plot】
9:可以得到HAMP在【LUSC】这个数据集中的P值和HR值,HR=1.14(0.9-1.45),logrank P =0.26,和文章中列举的数值一模一样。


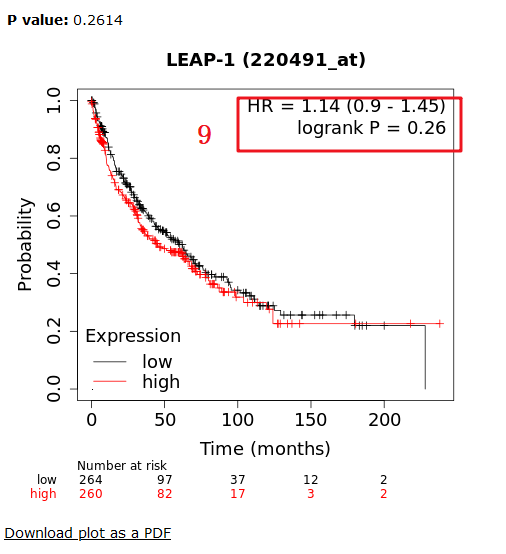
10:接下来我们就按照这个红框框住的选项,一项一项摘录数据,整理完毕后,上传到仙桃可以做(根据上传格式,只要格式正确,秒出图),也可以用Graphpad进行画图,这里就不过多介绍了。
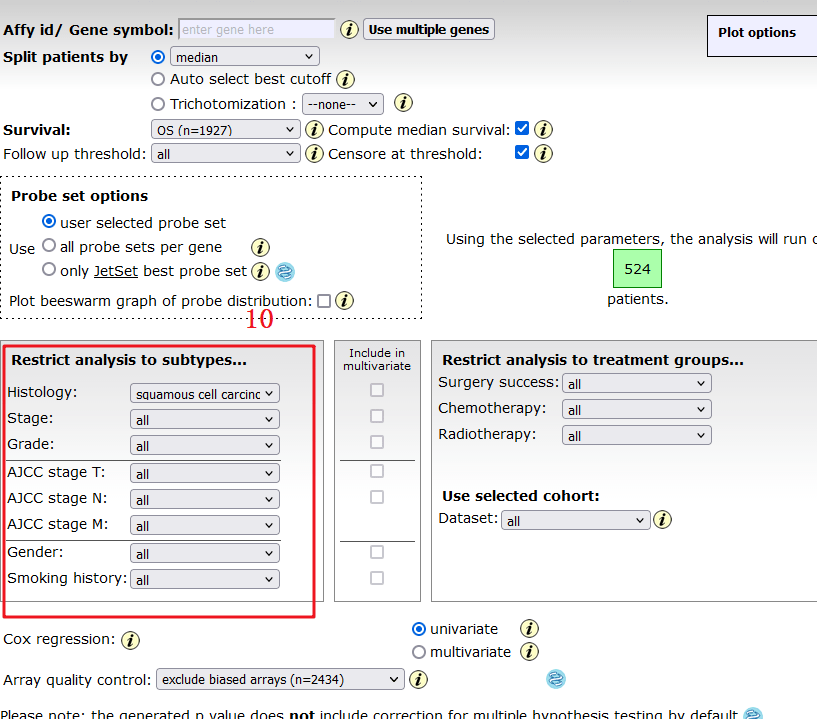
至此,Figure 4 完整复现完毕。
Figure 5 鉴定与Hepcidin互做的基因
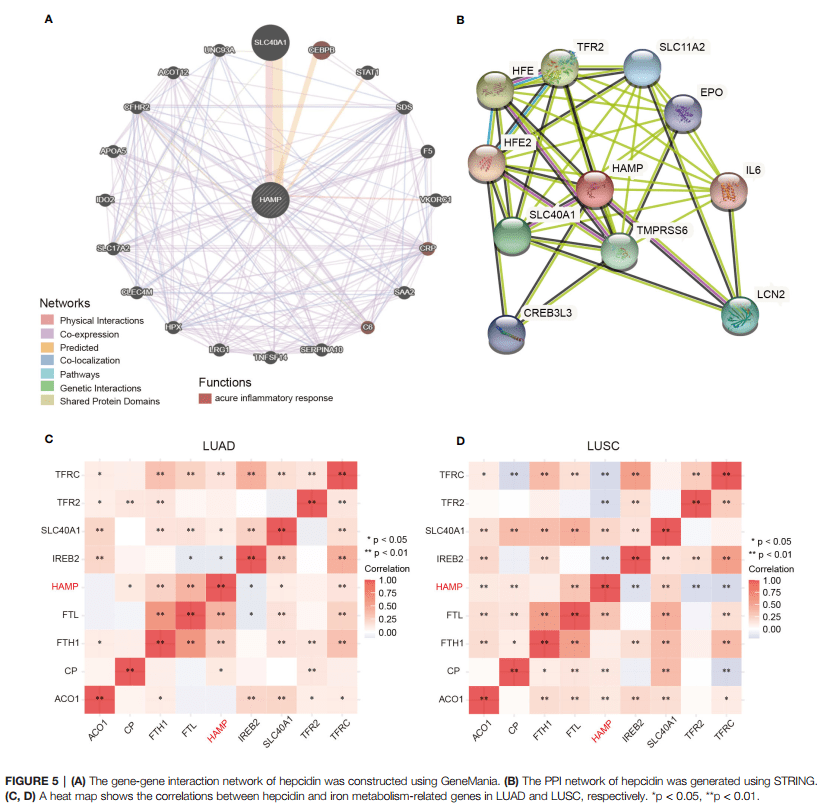
首先我们来复现Figure 5A:分析前20个与Hepcidin相互作用的基因。
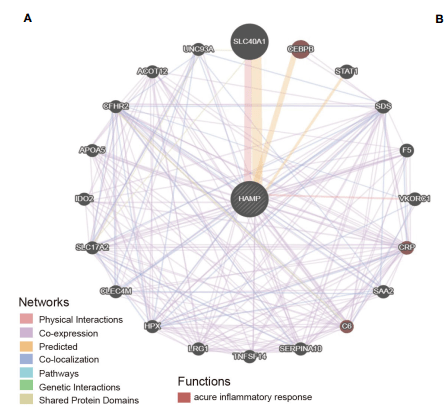
GeneMANIA这个数据库可以用来发现相关基因,包括蛋白-蛋白,蛋白-DNA和遗传相互作用,通路,生理生化反应,基因和蛋白表达,蛋白结构域及表型筛选,而且数据都是定期更新的,蛮好用的哦~
复现步骤具体如下:
1:打开GeneMANIA网址,http://genemania.org
2:输入我们需要研究的靶基因【HAMP】
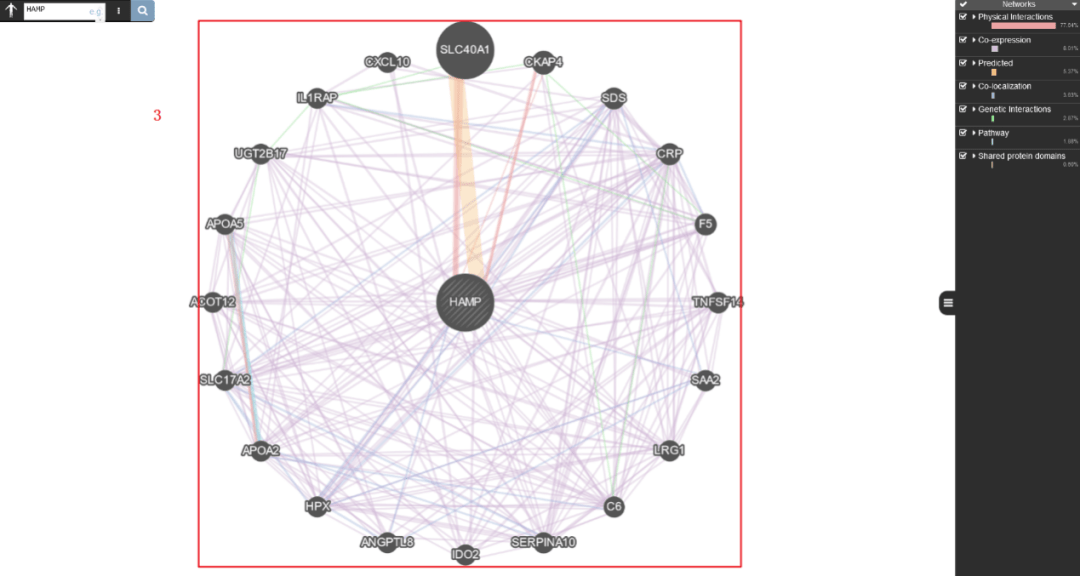
3:即可出现和靶基因相互作用的网络,排名前20的基因,和原文中Figure 5A 一模一样
接下来我们复现Figure 5B:string数据库分析相互作用的基因
复现具体步骤如下:
1:打开string网址
2:选择【protein by name】模块
3:输入研究的靶基因【HAMP】
4:选择物种【Homo sapiens】
5:点击【SEARCH】
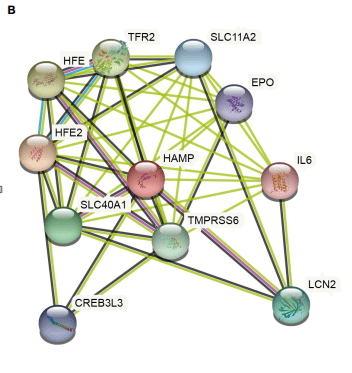
6:即可出现PPI互作网络,出来的20个相互作用基因,其中有17个相同,3个不同,原因可能是string数据库也是一直在更新的,导致这些许的差异,不过这不是重点,我们主要是学习思路与方法。图片保存下来即可得到Figure 5B。
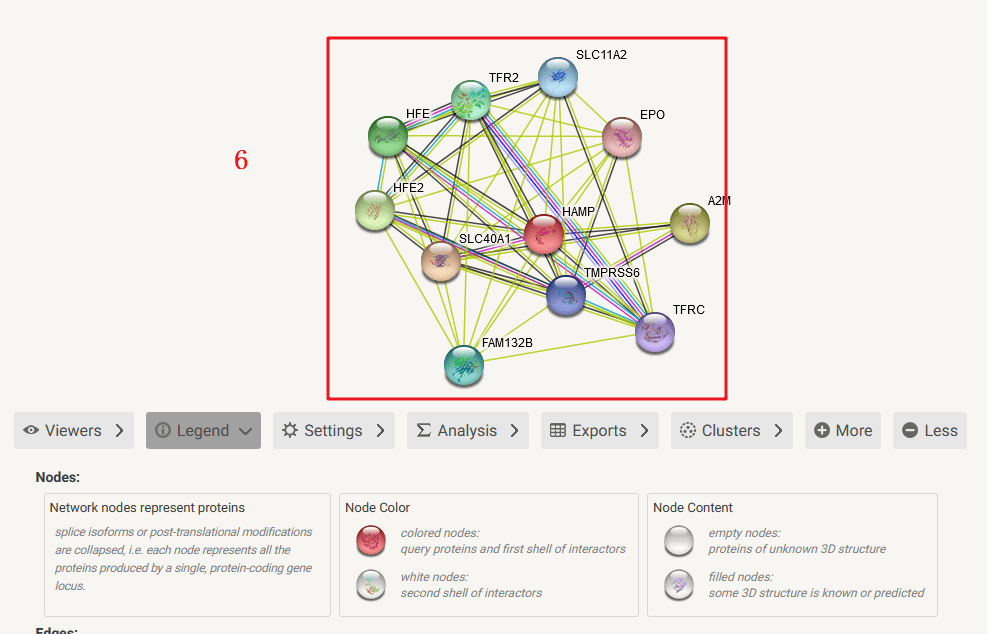
Figure 5C,D分析铁调素与铁代谢相关基因的相关性
由于作者用的是pheatmap R包做的,利用TCGA数据分析铁调素与铁代谢相关基因的相关性,我们可以选择仙桃生信工具进行复现, 基础版绘图就可以做相关性热图。
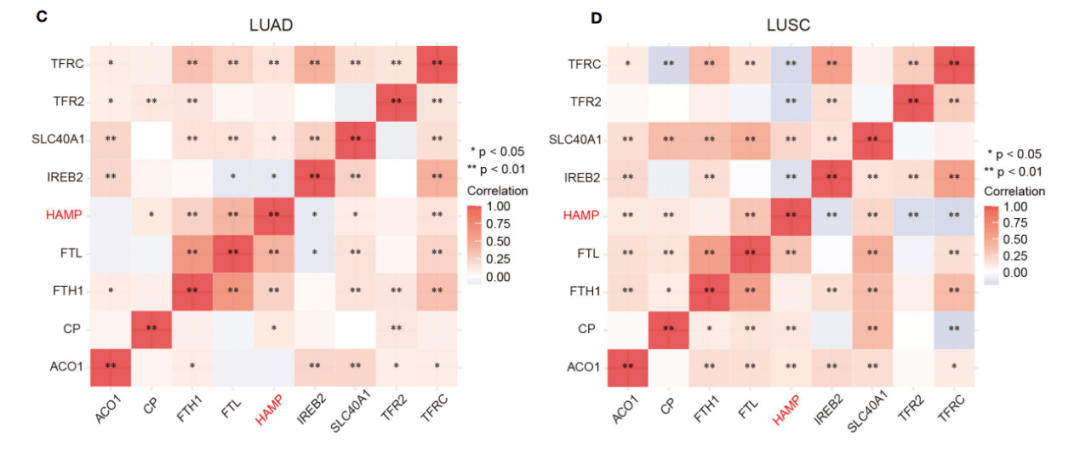
具体复现步骤如下:
1:进入仙桃工具,选择【基础绘图】【相关性热图】
2:上传示例数据这一步,由于我这里没有下载LUAD TCGA数据,所以给大家展示一下这种相关性热图需要的数据格式。
每一列代表一个指标,每一行代表一个样本,我们下载TCGA LUAD数据之后,提取这几个基因的表达值,就可以进行两两相关性分析,得到相关性矩阵后再进行可视化,会得到行名和列名都相同的相关性矩阵。
我们有3种样式供我们选择,分别是R包:corrplot的可视化结果
样式2:ggplot2包的可视化结果(本文就是利用这个包做的相关性分析)
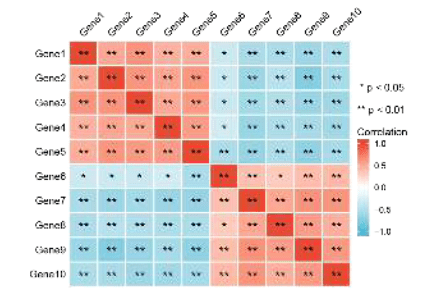
样式3:对角线热图是ggplot2可视化的另外一个结果

大家按自己喜好选择可视化方法,仙桃真是太香了有木有~
Figure 6:对差异基因进行富集分析
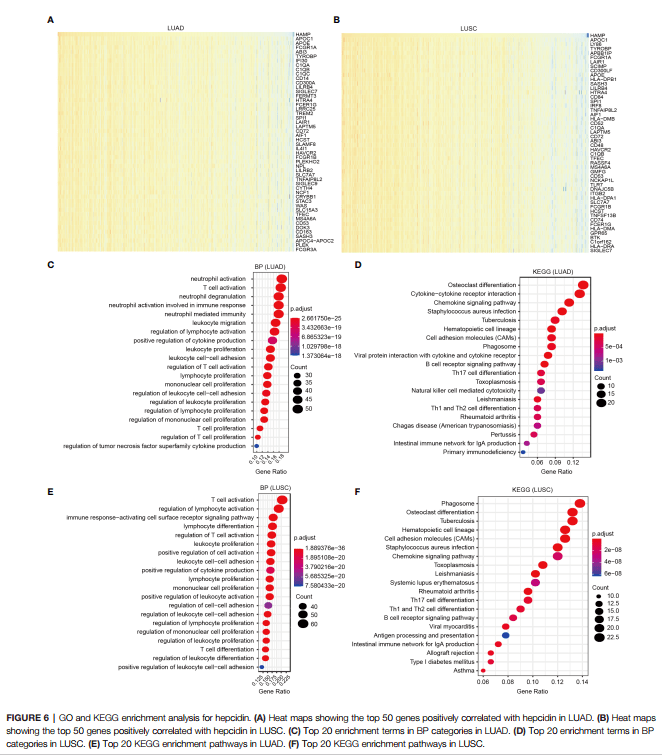
Figure 6A、6B是展示TCGA前50个差异基因热图
这个图说实话蛮丑的,接下来让我们用仙桃工具做【单基因公表达热图】因为大师兄说TCGA有这张图就完全够啦
具体复现步骤如下:
1:打开仙桃工具【交互网络(联)】
2:选择【分子相关性分析】
3:选择【单基因共表达分析】
4:选择研究的癌种【LUAD】
5:输入我们的目标基因【HAMP】
6:输入分子列表,这个列表最多能输入50个基因,正好和文章当中展示的基因数一样,但我们这里为了方便,我们输了前10个基因,分别是:APOC1 APOE,FCGR1A,ABI3,TYROBP,IFI30,C1QA,C1QB,C1QC,CD14,这样单基因共表达热图就做好啦,【LUSC】的同样的做法,大家可以自己试一下~
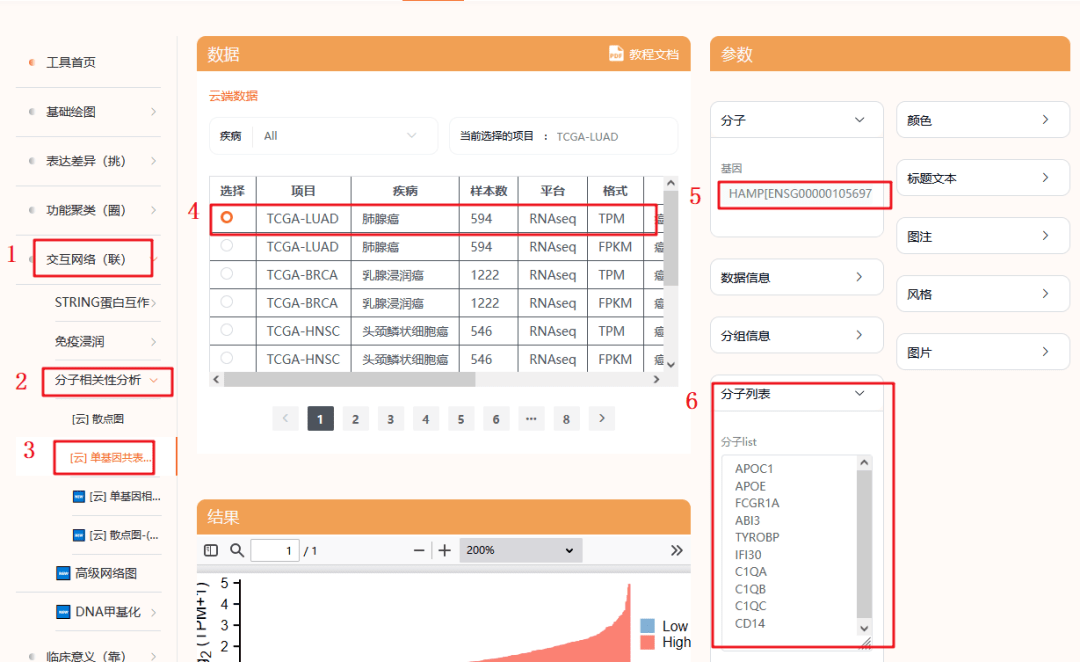
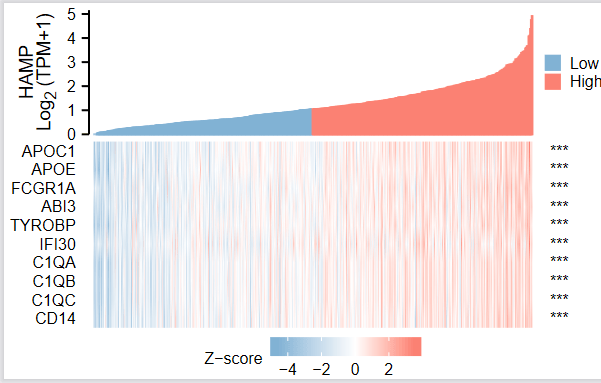
Figure6C,D,E,F 展示了GO和KEGG分析 。
作者在材料和方法中提到,GO/KEGG是利用 ClusterProfiler包做的,我们依然是利用仙桃学术工具来复现
所谓富集分析:简单而言,就是拿一堆有功能注释的分子与所有有功能注释的分子去比较(超几何分布检验),确定那一堆中的基因都涉及了哪些功能作用。(摘自大师兄的教程文档)

Figure 6A/B/C做的都是GO分析(包含了BP,CC,MF)
复现步骤具体如下:
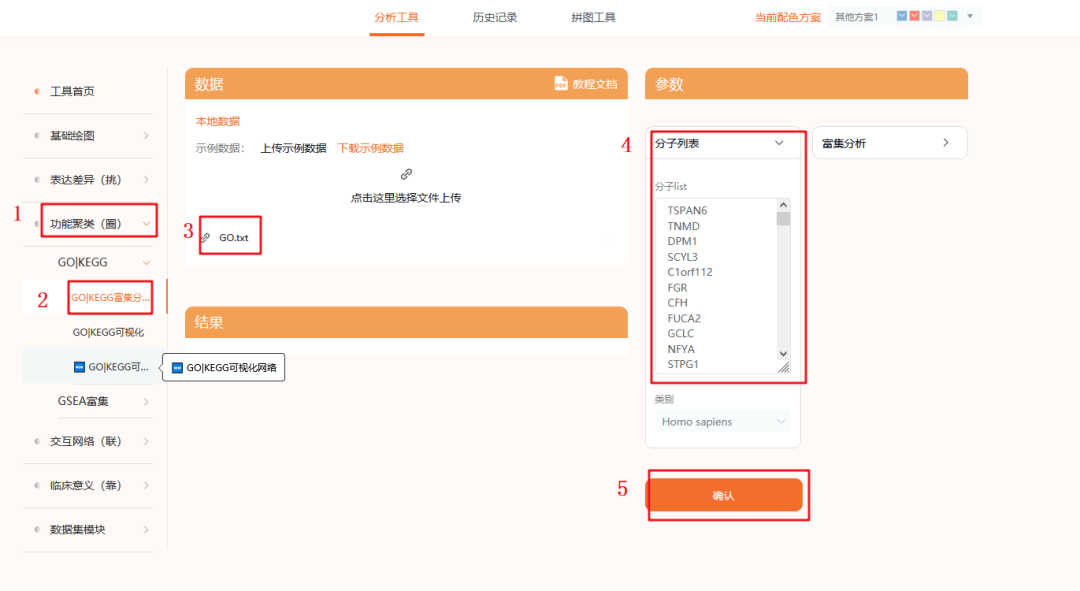
1:进入仙桃工具,选择【功能聚类】模块
2:选择【GO/KEGG】富集分析
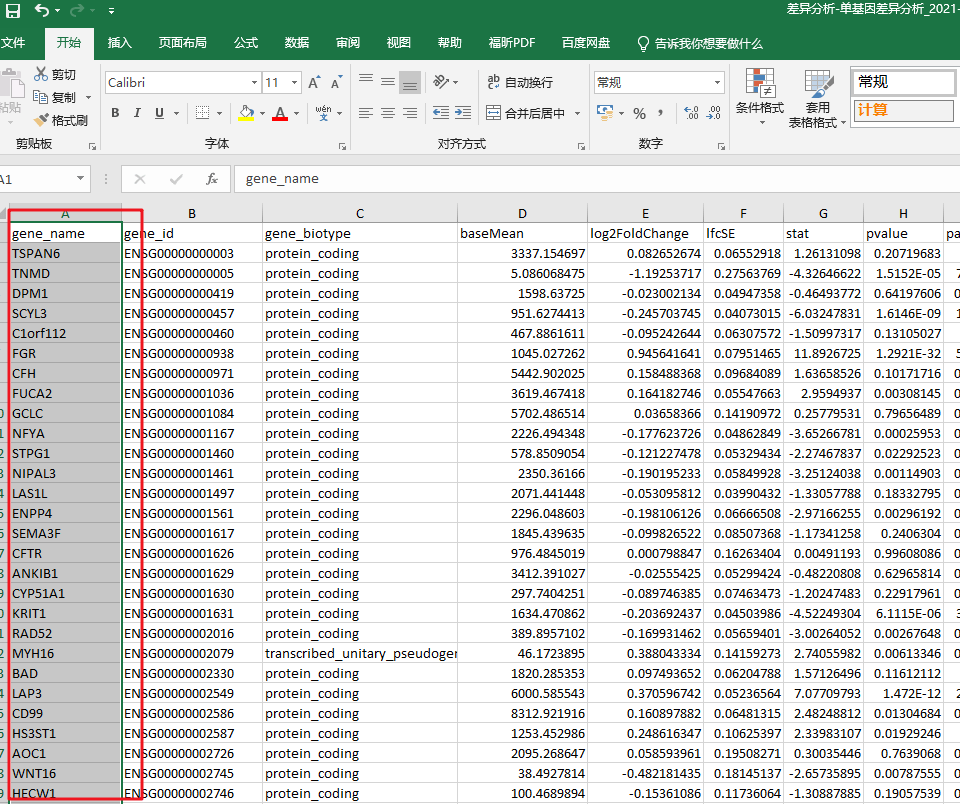
3:这里上传需要富集的差异基因列表,文章当中提到取了前300个基因做富集分析,我们就从单基因差异列表里复制第一列即可(做富集分析只需要第一列:基因名)
4:文件上传成功后,这里就会自动出现上传的基因列表
5:点击确认后,出现结果后点击保存,历史记录里就会出现我们进行富集分析的结果
然后我们进入【GO/KEGG可视化】模块
1:选择【GO/KEGG可视化】模块
2:选中我们之前保存的GO/KEGG结果
3:选择文章当中使用的气泡图
4:点击确认,下面出图,我们这里选了BP的前5个通路,文章当中列里20个,这个可以个性化设置,大家可以自己试试。
(因为输入想要可视化功能或者通路的数目,可以根据需要修改,而修改的方式是需要在历史记录中找到对应的记录,下载EXCEL结果,复制想要展示的ID到这个输入框中,一行代表一个)
剩下的CC,MF,KEGG都是一样的操作方式,大家可以自行试一下。
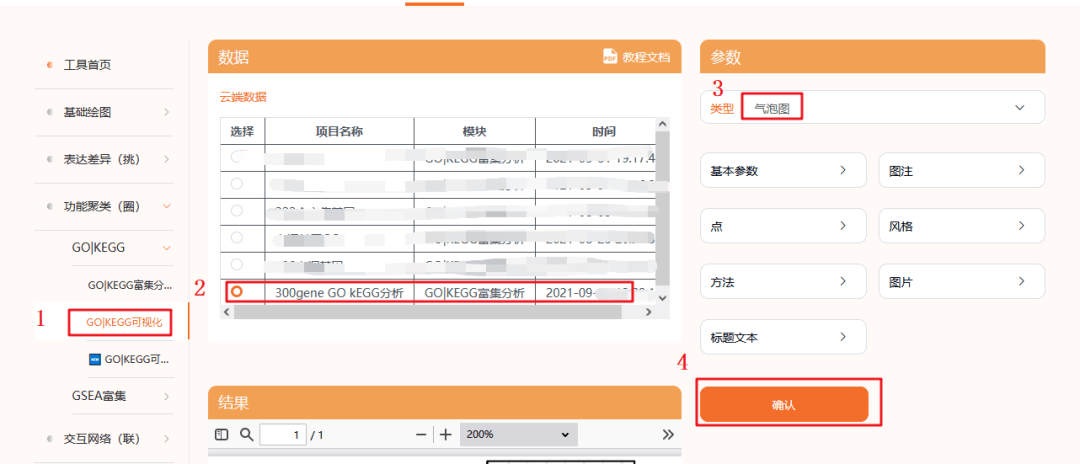
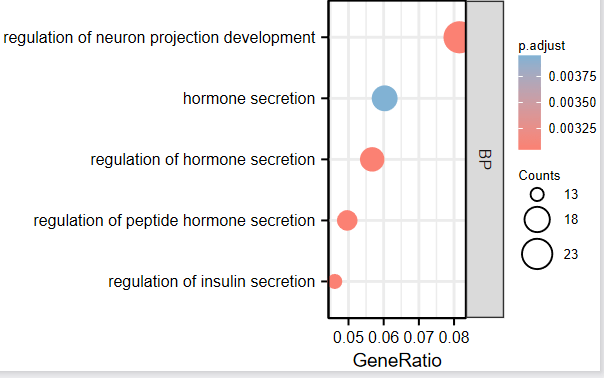
Figure 7:铁调素表达和免疫浸润细胞的相关性分析

我们先来复现Figure 7A:利用TIMER数据库分析免疫浸润情况
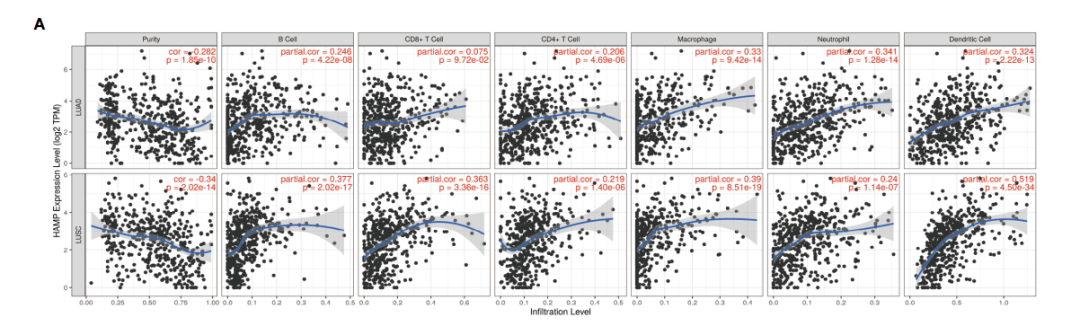
作者用了TIMER数据库,分析了在LUAD,LUSC中HAMP表达和肿瘤纯度及各免疫细胞之间的相关性,我们以LUAD为例
具体复现步骤如下:
1:进入TIMER数据库,网址为:https://cistrome.shinyapps.io/timer/
2:选择【Gene】模块
3:在【gene symbol】处输入分子【HAMP】
4:在【Cancer type】处,输入【LUAD】,随后换成【LUSC】即可
5:这里默认会加载6种免疫细胞,保持默认即可。
6:点击【submit】
7:如果网速好的情况下,几乎秒出图,我们对比了一下和原文一模一样,下载待用即可。
Figure 7A复现完毕。
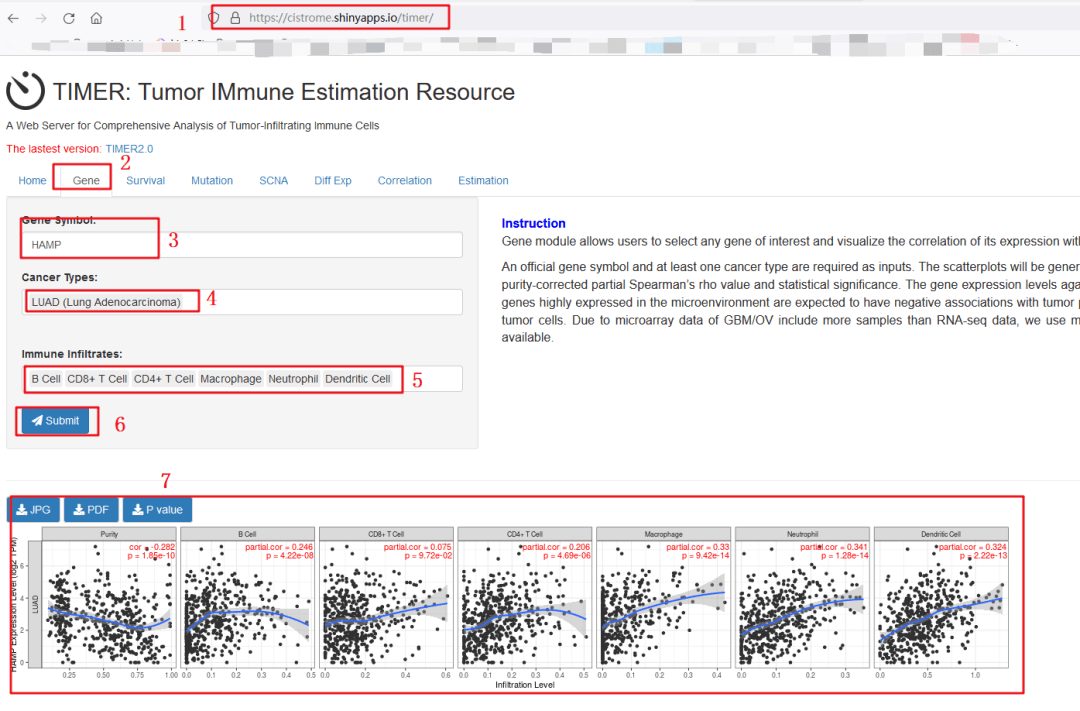
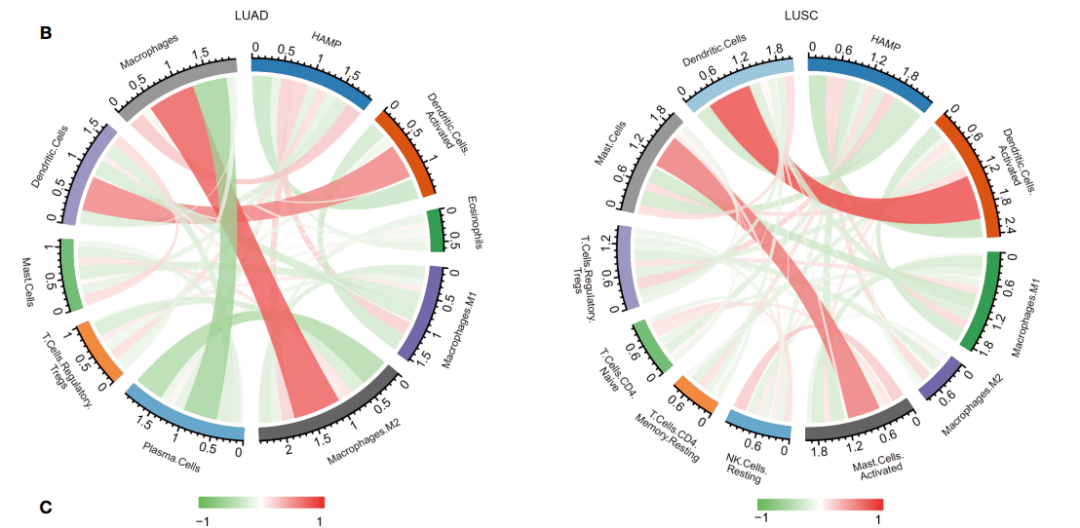
Figure 7B作者在原文中是利用cibersort计算免疫细胞浸润情况
原文中这个方法有些麻烦,我们可以利用ssGSEA,或者estimate包计算内置的markers计算样本的免疫浸润得分
基质得分和估计得分,一样可以分析免疫浸润的程度。让我们来尝试用仙桃工具来复现吧~绝对让你大呼“真香!
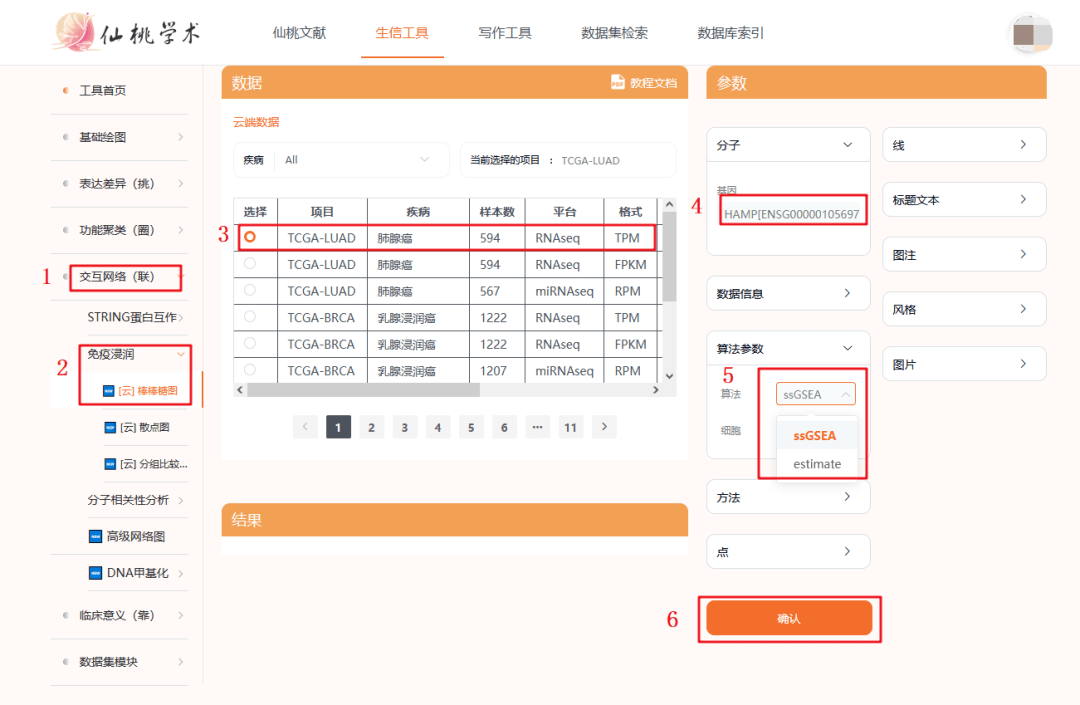
复现步骤如下:
1:打开仙桃学术工具,选择【交互网络】模块
2:选择【免疫浸润】--【棒棒糖图】
3:再分别选择癌种【LUAD】【LUSC】
4:输入靶基因【HAMP】
5:选择算法【ssGSEA】或者【estimate】
6:点击确认,即可出图
7:可以下载PDF格式
目前这种棒棒糖图用在文章里还比较少,这种形式表达免疫浸润审稿人肯定会有耳目一新的感觉,强烈推荐~保存LUAD】【LUSC】的棒棒糖图,相当于Figure 7B复现完毕。
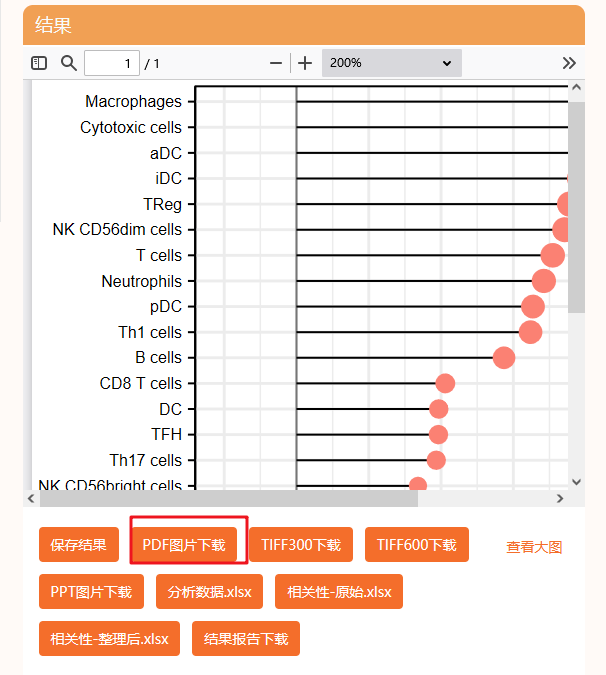
现在我们来复现Figure 7C:作者是将3个免疫检查点(CD274,PDCD1, CTLA4)和HAMP做了相关性分析。
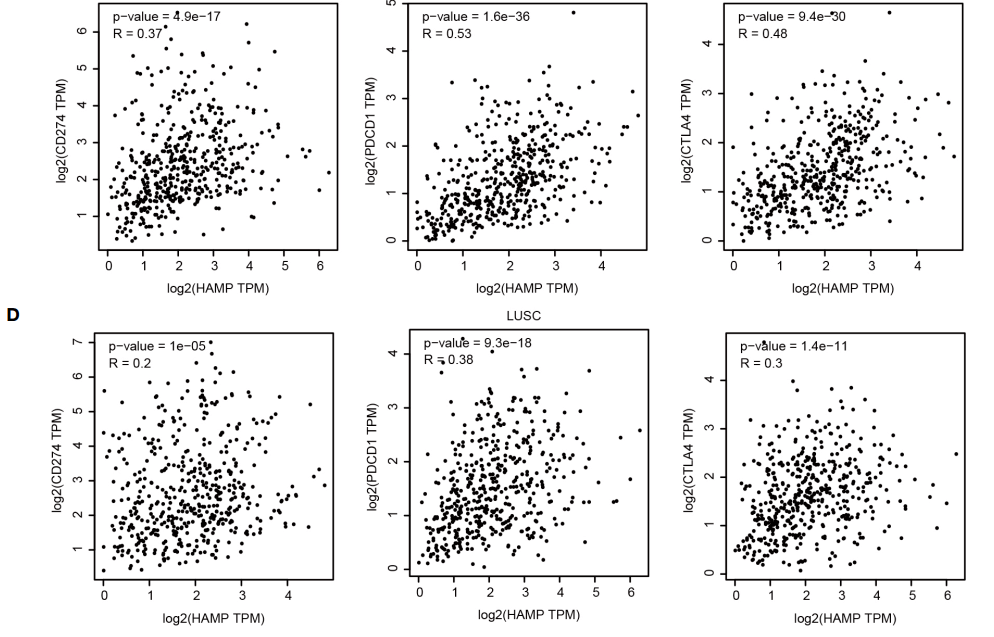
具体复现步骤如下:
1:打开GEPIA数据库,网址如下图所示
2:输入靶基因【HAMP】
3:点击【GoPIA】
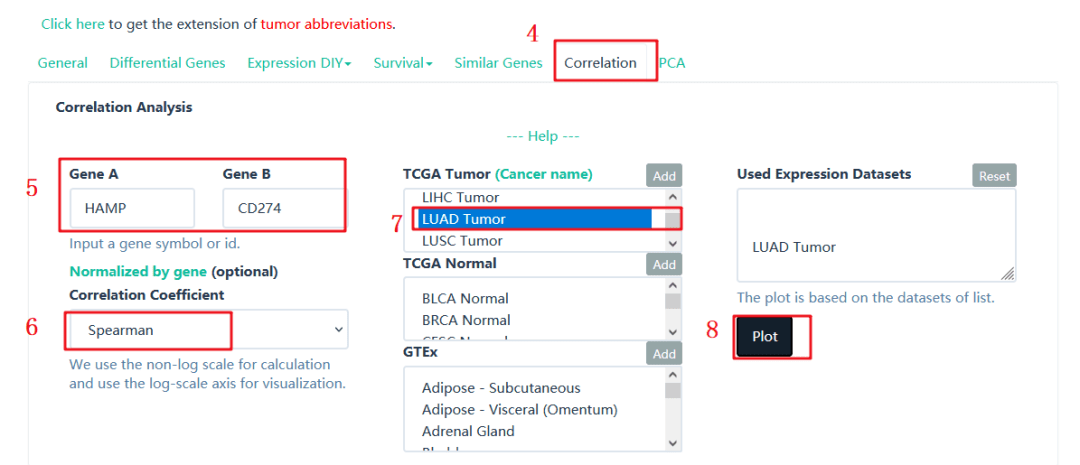
4:点击【Correlation】也就是选择了相关性
5:输入需要研究相关性的两个基因【HAMP】【CD274】
6:作者这里选了【Spearman】
7:分别选择【LUAD】【LUSC】
8:点击【plot】,即可出图,我们从途中可以得到信息【p-value】为4.9e-17,R=0.37
同样的方法,继续分析【HAMP】与【PDCD1】【CTLA4】的相关性,即可得到一模一样的Figure 7C ,7D
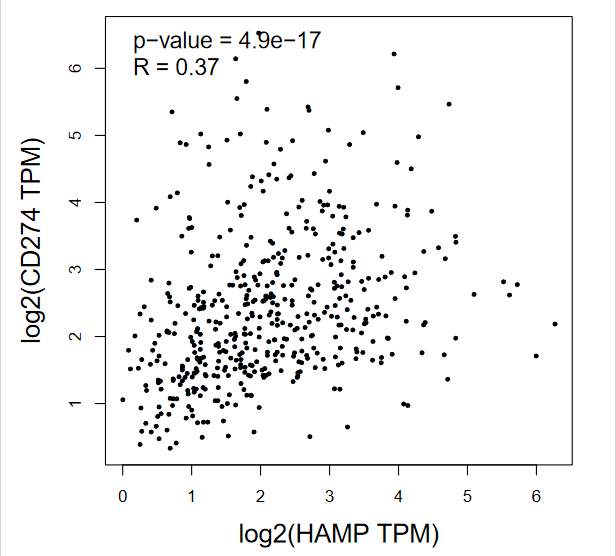
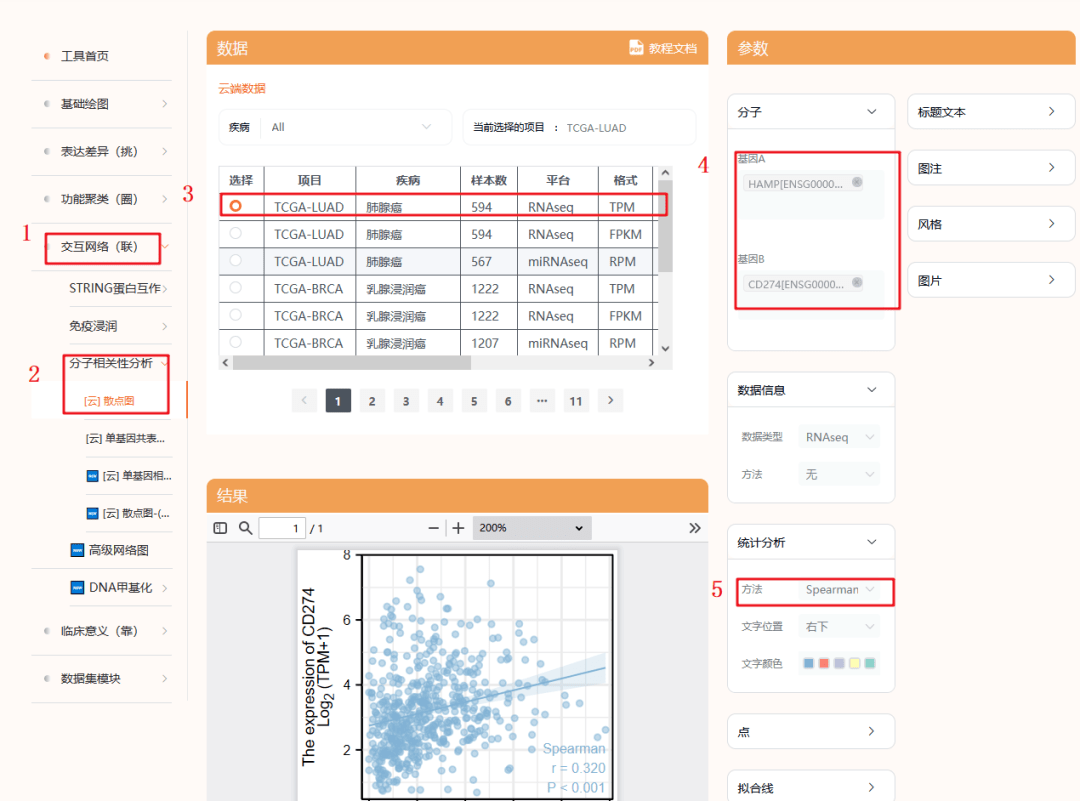
现在用我们仙桃工具来复现Figure 7C/D,分析靶基因和免疫检查点的相关性
具体复现步骤如下:
1:进入仙桃工具,选择【交互网络】模块
2:选择【分子相关性分析】--【散点图】
3:分别选择【LUAD】【LUSC】
4:输入需要研究相关性的两个基因【HAMP】【CD274】
5:我们这里的统计学分析方法依然选择【Spearman】
6:点击【确认】即可出图
工具的优势在于散点图的颜色可以根据我们自己的审美进行调整,不易与其他文章撞色,新颖性提高了一大截。
同样的方法,继续分析【HAMP】与【PDCD1】【CTLA4】的相关性,即可复现Figure 7C/D,2种方法,任君选择。
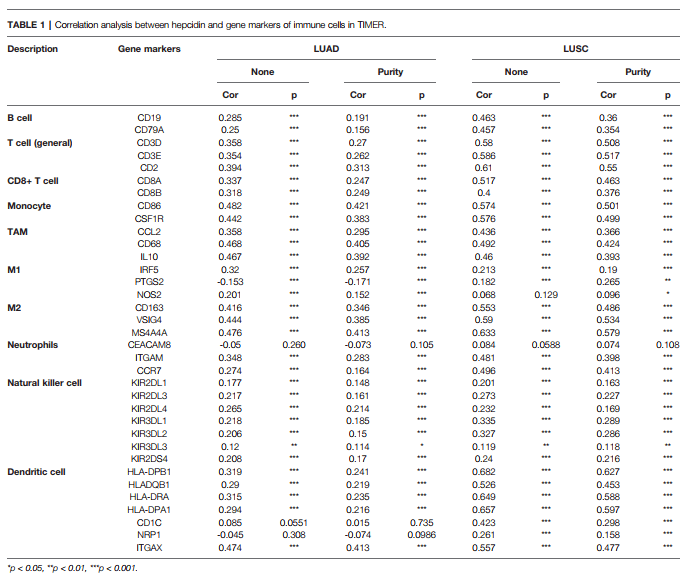
接下来我们来复现Table 1:利用TIMER数据库分析铁调素的表达和gene marker的相关性
复现具体步骤如下:
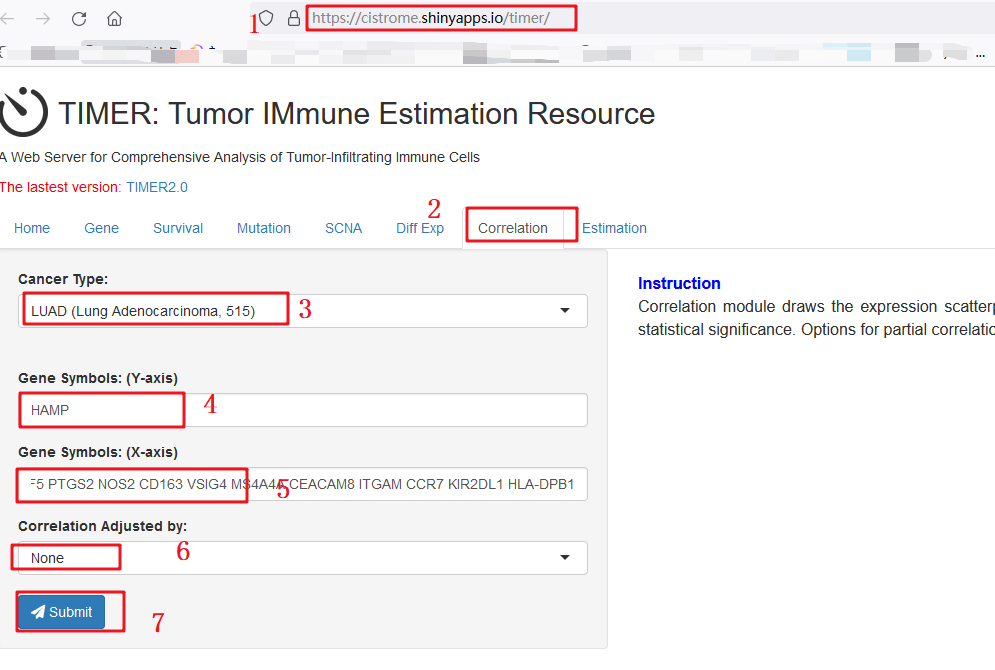
1:我们先来观察一下Table 1,作者分析了【LUAD】【LUSC】两种癌HAMP的表达和免疫marker的相关性。我们主要以复现【LUAD】为例。首先,打开TIMER数据库
2:选择【correlation】模块
3:输入我们研究的癌种【LUAD】
4:输入gene symbol【HAMP】
5:输入我们需要研究相关性的gene marker,分别是CD19 CD79A CD3D CD3E CD2 CD8A CD8B CD86 CSF1R CCL2 CD68 IL10 IRF5 PTGS2 NOS2 CD163 VSIG4 MS4A4A CEACAM8 ITGAM CCR7 KIR2DL1 HLA-DPB1
6:在这一模块中,我们先选择【None】然后选择【Tumor purity】
7:点击【submit】
8:下方出图,直接把相关性和P值直接标出来了,我们也可以直接下载表格,直接可以放在文章当中,这样Table 1复制完毕。
同样Table 2也是和Table1同样的方法。
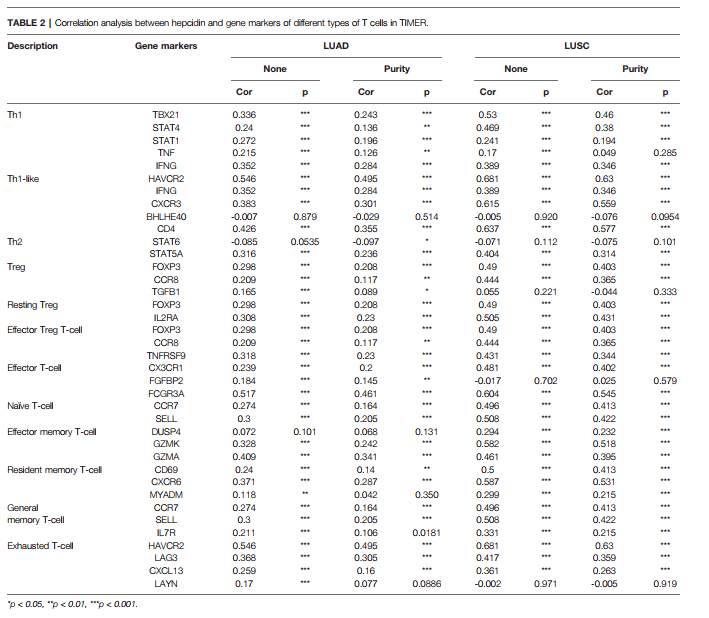
作者这里列举了HAMP和不同类型T细胞的基因marker的相关性,具体复现步骤如下:
1:我们先来观察一下Table 2,作者分析了【LUAD】【LUSC】两种癌HAMP的表达和T细胞的基因marker的相关性。我们主要以复现【LUAD】为例。首先,打开TIMER数据库
2:选择【correlation】模块
3:输入我们研究的癌种【LUAD】
4:输入gene symbol【HAMP】
5:输入我们需要研究相关性的gene marker,分别是TBX21 STAT4 STAT1 TNF IFNG CXCR3 BHLHE40 CD4 STAT6 STAT5A FOXP3 IL2RA FOXP3等
6:在这一模块中,我们先选择【None】然后选择【Tumor purity】
7:点击【submit】
8:下方出图,直接把相关性和P值直接标出来了,我们也可以直接下载表格,直接可以放在文章当中,这样Table 2复制完毕
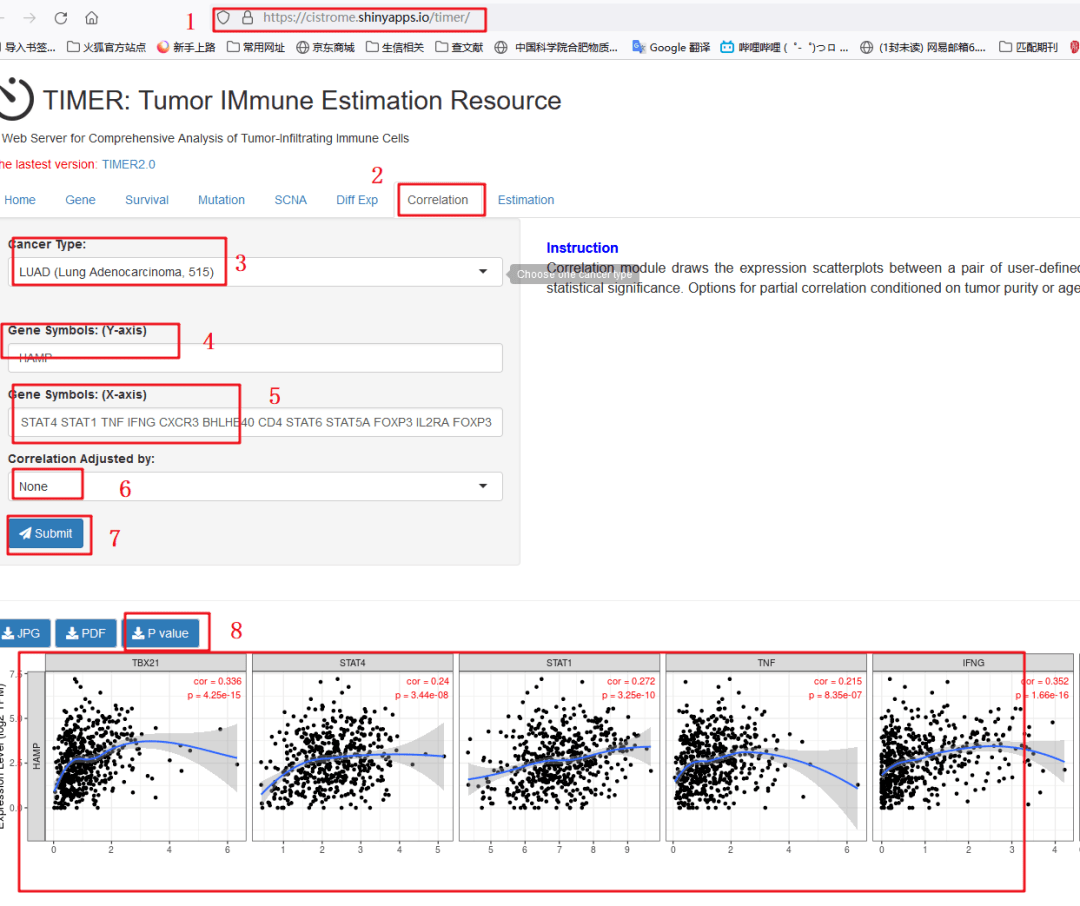
Figure8:基于LUSC患者免疫细胞的铁调素表达的预后分析
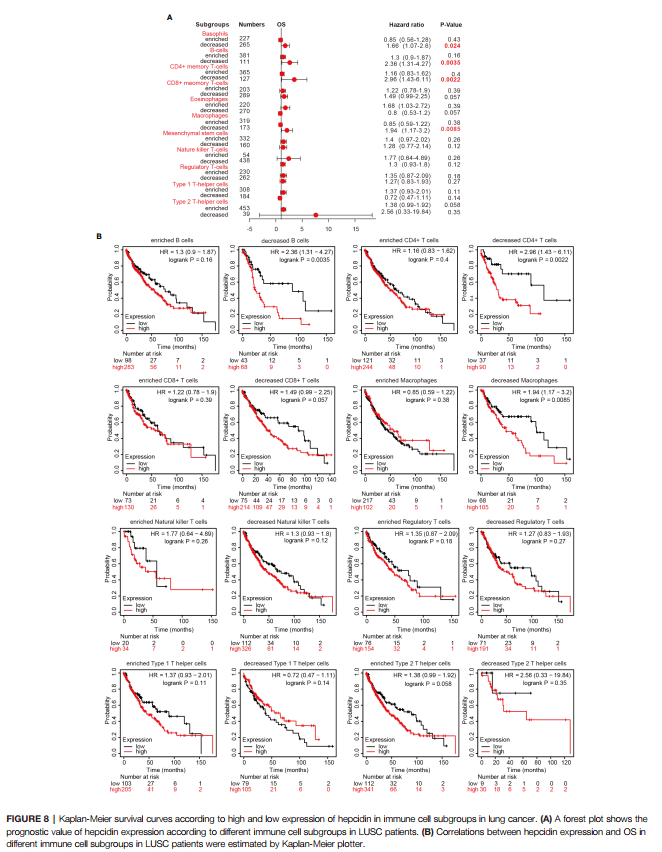
作者通过前面的分析得到H AMP和预后,免疫浸润相关,那作者就想,H AMP影响L USC的预后是因为免疫浸润的缘故吗?
所以作者在设定免疫细胞存在与否的条件下,再次用k m plotter数据库对肿瘤的存活率进行深度分析,很值得我们学习。
仔细观察F igure 8A-B,我们可以知道这两张图其实展示的是一个意思,只是通过F igure8B的生存分析,摘录HR 值,P值后,再将这些数据整理成森林图,所以我们先来复现Figure 8B。
复现具体步骤如下:
1:进入km-plotter数据库后,一定要记得选的室pan-cancer RNA-seq
2:输入我们研究的靶基因【HAMP】
3:选择作者研究的癌种【LUSC】
4:我们以Figure 8B的第1,2张小图为例,原文中展示了B细胞enrich和decreased两种状态,我们在这一步点击下拉菜单即可选择
5:点击【Draw-Kaplan-Meier plot】即可出图
和原文一模一样,我们将第一张小图的H R=1.3(0.9-1.87),l og-rank p=0.16摘录下来,整理成仙桃工具可以识别的格式上传。即可出森林图,这个功能仙桃工具的基础版即可作图。下面简单展示一下。

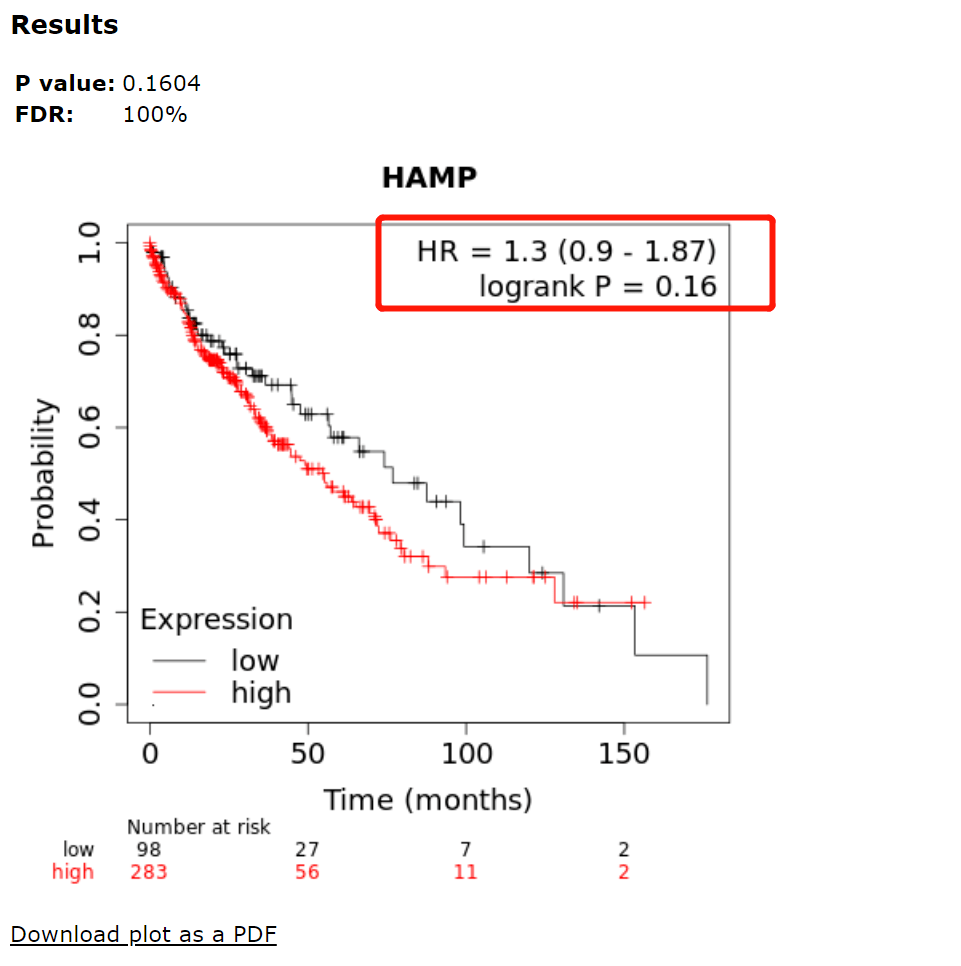
将Figure 8B每一张小图的HR值和P值摘录下来以后,我们整理成如下的格式就可以上传。
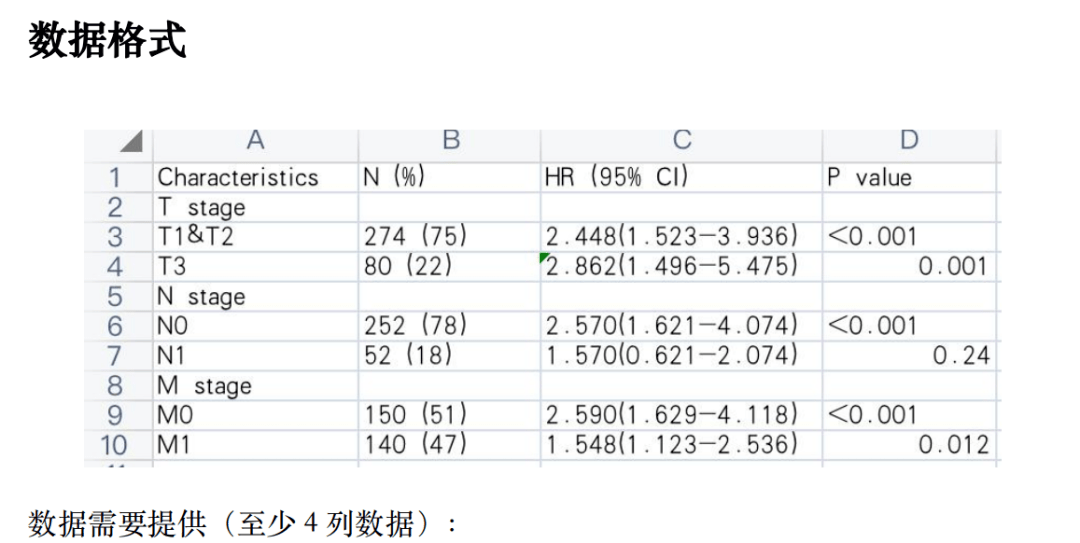
整理好之后,进入仙桃工具页面,点击【基础绘图】,选择【森林图】,然后上传自己的数据,点击确认即可出Figure 8A啦,小伙伴们快用自己的数据试一试吧~
最新的二区7+SCI复现太厉害了吧?感觉好有压力
看这里,减压福利来了~~手把手带你文章复现
转自:解螺旋- 本文固定链接: https://maimengkong.com/zu/1527.html
- 转载请注明: : 萌小白 2023年5月13日 于 卖萌控的博客 发表
- 百度已收录
