
大规模并行测序技术或下一代测序已成为基因诊断和研究的标准技术,尤其是外显子组和基因组测序现在已经在世界范围内广泛应用于患者的分子诊断。在过去几年中,许多实验室都在努力应对基于全新技术建立基因检测工作流程的挑战。测序技术中持续引入新的仪器、化学和分析方法加剧了这些挑战。
在过去十年中,新的测序技术已经上市,而其他技术已经消失,并且所有这些技术都经历了快速的变化和升级。外显子组捕获试剂盒、配套设备和耗材也是如此。在这个不断变化的领域,实验室一直致力于生成高质量的测序数据。
各种研究报告了测序数据中的偏差如何导致外显子组和基因组测序的敏感性降低或假阳性变异。例如,对于NGS 而言,高测序错误率和PCR 重复将导致潜在的假阳性calls ,而不均匀的序列覆盖或缺乏覆盖可能会导致灵敏度降低。其他问题,如strand 偏倚和插入大小分布也可能对测序结果产生不利影响。NGS技术比传统的基因检测方法更加数据密集,需要信息技术(IT)和生物信息学方面的专业知识,而这在许多实验室最初是稀缺的。生物信息学已经解决了为测序数据建立严格质量控制的困难,但也解决了从测序数据中可靠识别变异的挑战。例如,检测插入和缺失、识别短片段扩增重复或低覆盖区域的变异或区分单核苷酸变异(SNV)与测序错误相对困难。
此外,从外显子组数据中检测拷贝数变异(CNV )已成为一种标准程序,并带来了自身特殊的挑战。同样,与测序仪器一样,生物信息学需要处理来自软件工具、基因Panel 和其他注释资源的持续更新,以确保分子遗传学家拥有用于解释最新数据的最新信息。这反过来要求实验室实施自动测试其分析的策略以及重新分析现有数据的系统方法。
在新的测序可能性以及许多疾病的遗传和表型变异的推动下,临床基因检测在过去十年中发生了巨大的变化。根据临床表型,只有一个或几个基因会被测序;从靶向基因测试来看,现在通常涉及对大量疾病基因的分析。与单基因分析相比,外显子组或基因组测序中的大量变异的解释明显不同。这不仅需要对该技术有深入的了解,以便评估数据质量和已识别的变异,还需要新的变异解释方法。
NGS 变异的初始报告有时过于严格,从而忽略了与患者表型不完全匹配的变异,或过于宽松,导致许多意义不确定的变异(VUS )。随着时间的推移,测序数据的质量有了很大的提高,并且开发具有不同变异频率的大型公共可用数据库,如GnomAD 数据库,极大地帮助开发了更高效的变异筛选选项。此外,在过去的几年中,已经开发出各种建议和质量评估方案指导NGS 变异的解释、分类和报告。
现在有一些关于NGS 测试的指南可以帮助NGS 测试设计、优化、验证、质量管理和生物信息学等方面。尽管如此,仍然存在许多挑战,错误肯定会发生,即使在质量至关重要的受监管临床基因检测实验室也是如此。这里我们展示了我们实验室在十年临床外显子组测序过程中犯下的一些错误的例子,以及我们从这些错误中吸取的教训(补充表S1 )。虽然湿实验室有其特殊的挑战,但在这里,我们主要关注与数据分析和变异解释相关的问题。 我们希望通过分享这些例子,其他实验室可以避免犯同样的错误。
数据分析
对于许多诊断实验室来说,数据管理和测序数据分析流程的开发已经变得非常重要。构建一个完整、高效和稳健的NGS 分析流程是一项复杂的任务,包括多个微妙的步骤,包括从NGS 读取的比对到不同类型遗传变异的调用和注释,如SNV 、小插入和缺失、CNV 和短串联重复序列(STR )。由于需要执行许多不同的处理步骤,并且数据量很大,因此相对容易犯一个小错误,对最终结果产生较大但不明显的影响。在这里,我们展示了自己在数据分析过程中犯下的五个错误的例子,这些错误到目前为止还没有在文献中得到充分强调。
1. 序列质量
“垃圾输入,垃圾输出”是计算机科学中的一句名言。它抓住了一个概念,即有缺陷的输入数据会产生有缺陷的输出或“垃圾”。这同样适用于测序数据。我们的实验室在测序结果方面遇到了许多问题,这些问题不是由于数据处理中的错误,而是由于初始数据生成本身存在问题。确定下游问题的根本原因可能是一项具有挑战性的任务,因为测序数据中的细微质量问题可能会对后续突变检测产生很大影响。一个相对常见的问题是数据中有许多虚假的变异,这种情况有时是由于意外的高测序错误率、样本污染,或由于adapter 序列的不正确修剪(补充图S1 )。

大多数质量问题可以通过检查原始测序数据或变异的质量分数较低,并且偏离杂合子突变丰度50% 来识别。相反,在大多数情况下,变异的数量减少是由于序列覆盖率低。然而,灵敏度降低可能还有其他原因。在两批外显子组测序样本中,我们注意到变异的数量较少,只是因为我们对几批样本进行了趋势分析。最初,我们预计这是由于样本的序列覆盖率较低(补充图S2 ),然而,这些样本的序列覆盖率与其他样本没有区别,最终我们发现这个问题是由于duplication reads 的比例增加了10-20% 。由于duplication reads 可能是由于PCR 扩增并可能引入假阳性变异。大多数变异的分析软件不会考虑它们的变异调用。因此,许多区域的有效覆盖率比这两个批次的有效覆盖率低10-20% (补充图S2 )。许多质量问题可以通过使用Qualimap 等工具轻松识别,这些工具可以计算测序实验的质量统计数据,如覆盖率统计数据、测序错误率和重复读取的百分比。
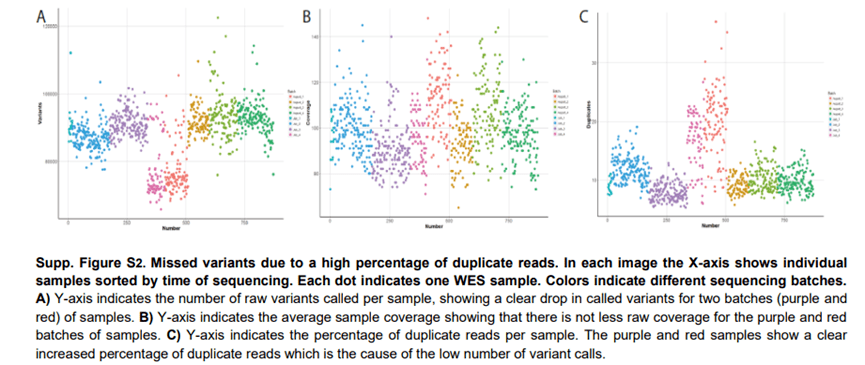
因此,我们强烈建议在生物信息流程的所有步骤中嵌入广泛的质量控制,并遵循质量参数的趋势,如重复读取的百分比、覆盖率分布、变异总数以及在gnomAD 中未发现的罕见变异的百分比。应密切调查与预期值的偏差。在开发和测试期间确定质量阈值将有助于以后识别质量问题。当实验室协议发生变化时,例如随着新测序仪器的引入,这些阈值可能需要更新。对测序数据进行全面的质量控制分析可以防止数据解释的许多下游问题。
.
2. 序列比对:alternate contigs
NGS 数据最主要的处理步骤是将读取数据与参考基因组对齐。然而,特定区域的基因组结构在不同的个体和群体之间可能存在很大差异。为了正确地表示这些位点,参考基因组使用alternate contigs ,即基因组中特定区域的不同参考序列。这些交替重叠群包含基因组中的区域,这些区域以如此复杂的方式变化,以至于它们无法表示为单个参考序列。在我们最初的分析工作流程中,我们试图尽可能全面,包括可能最大的参考基因组,其中包括alternate contigs 。然而,默认情况下,大多数reads 都会给与参考基因组中多个区域一致的比对质量分数。这些比对质量(MAPQ )等于零的读数通常在集成基因组学查看器(IGV (Robinson 等人,2011 ))中以空白读数显示(图1A )。变异检测算法反过来会忽略这种读取,并且不会在读取MAPQ 分数较低的区域识别变异。这些区域变异虽然可以通过手动检查看到,但不会被分析。这一错误是在实验室专家的帮助下确定的,他们查看了比对后的测序数据,以确定隐性基因中是否存在潜在的第二个突变(见VI-3 )。我们发现通过包含alternate contigs 读取无法明确对齐的编码基数将增加三倍。
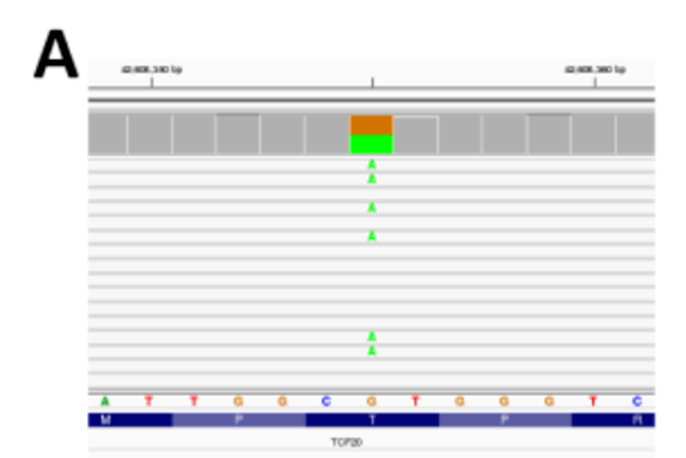
图1A
最近,英国生物银行(UK Biobank )的数据也报道了同样的问题,在GRCh38 参考基因组中引入了大量的替代性重叠群,导致数千种变异的缺失。有两种方法可以避免这个问题。简单的解决方案是在分析中简单地排除替代重叠群,这是目前我们在GRCh37 外显子组分析中所做的。在没有交替重叠的情况下分析数据,将正确对齐人类参考基因组主要组件中的reads (图1B )。一个更复杂的解决方案是应用对齐算法,可以使用相应的索引文件处理交替重叠,我们现在使用参考基因组的GRCh38 构建分析基因组。考虑到GRCh38 极大地扩展了替代性接触基因的种类(以及其他改进),行业开始向GRCh38 过渡将是有利的,以便能够正确地检测和分析群体特异性单倍型中的基因组变异。
3. 变异检测:捕获目标文件
有许多不同的外显子组试剂盒,它们都使用自己对“ 感兴趣区域” 的定义。从外显子组检测变异的最初方法是分析整个全基因组,而不考虑捕获目标或编码区域。然而,这在计算上很繁重,结果数据将包含许多来自不感兴趣区域中非目标读取的低质量变异。因此,将分析限制在可以合理预期可靠变异的充分覆盖范围区域似乎是合理的。尽管最初的外显子组试剂盒试图精确定位编码区,但许多制造商开始移动捕获探针,使其部分重叠或接近感兴趣的外显子,以优化富集效率。这背后的想法是:结合序列读取长度(通常为100-150 bp )和基因组DNA 片段的富集(延伸到目标之外但与目标重叠),不仅可以充分覆盖捕获目标本身,还可以覆盖100-150 个相邻碱基。这确实提高了许多“ 困难” 外显子的捕获效率,但使决定在哪些区域检测变异变得更加困难。
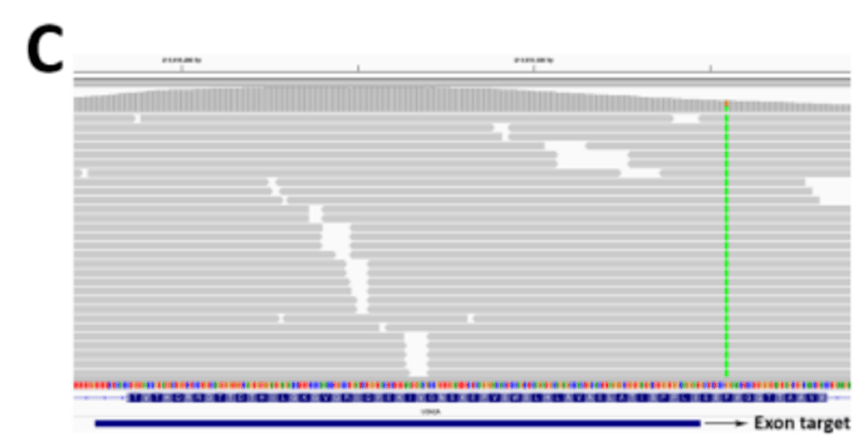
图1C
在我们最初实施新的外显子捕获设计时,我们犯了一个错误:只在外显子捕获目标区域中检测变异,没有意识到任何捕获目标都没有直接覆盖一部分外显子,因此遗漏了相关的编码变异(图1C )。尽管我们在测试外显子组试剂盒时进行了几次质量检查,但我们并没有立即意识到我们丢失了多达5.4% (1897KB )的所有编码区域(安捷伦SureSelect 版本4 )。同样,当通过IGV 在序列比对中发现可见的变异在vcf 文件中不存在时,观察到了这个错误。在最近的外显子组试剂盒中,与捕获目标相邻的编码碱基数量较少,但仍然相当可观(图1D )。
大多数制造商保证在捕获目标附近有足够的覆盖100bp ,但我们目前将目标扩展为200bp ,以平衡额外的计算时间和编码区域中的额外变异。显然,在全基因组范围内分析变异将绕过这些问题,但我们已经判断,额外的计算时间和低质量变异的增加并没有使这足够值得。我们估计,在全基因组范围内调用变异将使分析时间加倍,并将产生更多的变异, 其中一个重要部分是假阳性位点。在实施新的外显子组捕获设计时,强烈建议事先定义临床目标或感兴趣的区域,然后确定这些区域覆盖的完整性。
4.Exome CNV 分析:参考对照组
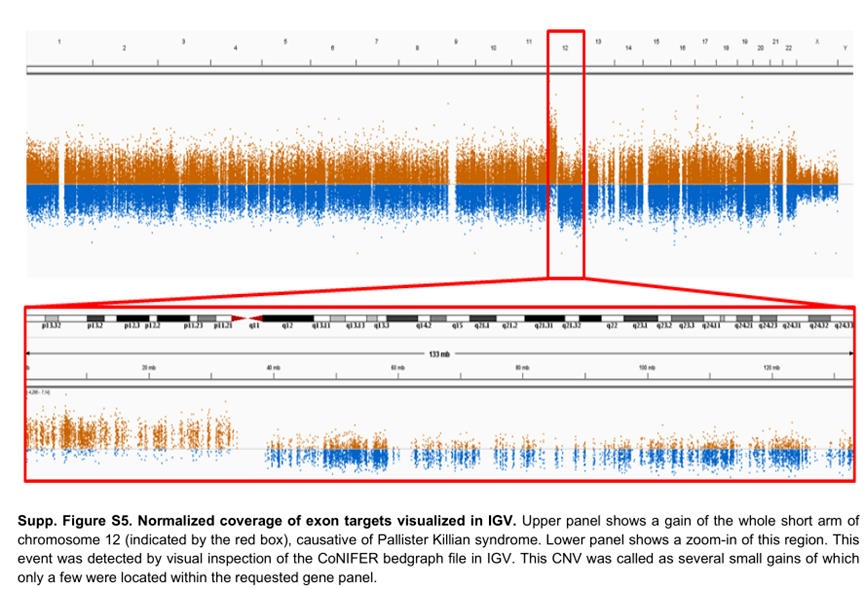
很早以前,人们就清楚WES 还可以根据样本之间序列覆盖深度的差异来推断CNV 。由于序列捕获和GC 含量,单个目标的覆盖率偏差阻碍了外显子组之间覆盖深度的比较。从外显子组数据中检测CNV 的大多数工具依赖于创建参考对照,以标准化每个区域的覆盖深度,并克服数据中的覆盖偏差。我们发现参考对照的大小和质量对CNV 的质量有很大影响。具有少量样本或具有不同测序特征的样本混合的参考对照,将导致测序目标预期覆盖率的变异性增加(图1E )。
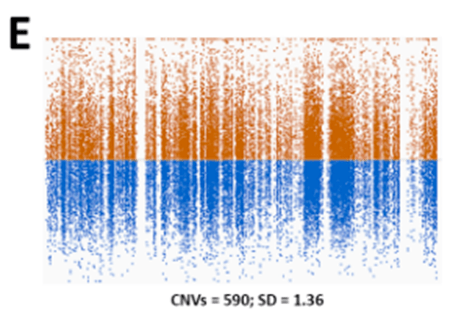
这将导致许多虚假的CNV ,使解释更加困难。2016 年,我们意外地在同一参考池中组合了使用两种不同方法比对reads 的样本。出乎意料的是,这不仅导致了虚假的CNV 被检出,还导致了大型CNV 被错过,但在之前的CNV 分析中已经检测到。目前,我们的CNV 参考对照使用最新样本不断更新,以使测序化学和方案的变化导致的技术变化最小(图1F )。
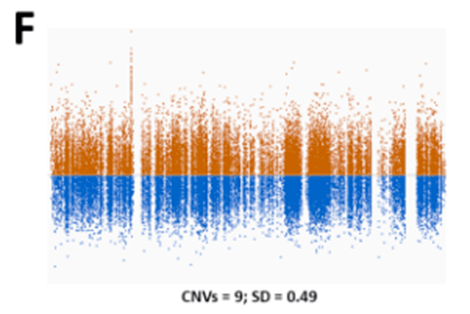
除此之外,根据测序平台、富集平台、在X 染色体上分析CNV 的性别,还使用了几个独立的参考池。为了了解潜在的质量问题,我们在趋势分析中监控每个样本和测序批次的CNV 呼叫数量,以及每个样本的标准化目标覆盖率的平均变异性。根据我们的经验,我们建议使用与捕获试剂盒、测序仪器和化学以及性别相匹配的CNV 参考对照。
5.注释:基因定义
虽然我们定期更新参考数据集,如人口频率、OMIM 信息和HGMD/ClinVar 分类,但我们最初没有定期更新我们的基因定义,天真地期望人类基因组中的所有基因和转录本都已被彻底绘制出来。基因定义是解释基因变异最基本的资源。有几种可用于基因定义的公开资源,例如RefSeq (由国家生物技术信息中心(NCBI )开发)和GENCODE ,它们结合了HAVANA 集团的手动注释和Ensembl 的计算注释。
有点让我们惊讶的是,当我们将2017 年GENCODE 基本基因定义更新为更新版本时,我们遇到了几个最初被注释为非编码的变异,但其结果是在一个新注释的外显子中,从而可能完全改变解释,例如基因CCDC141 (图1G )。
RefSeq 和GENCODE 仍有定期更新,这些更新会改变已知的基因定义,并对WES 变异的解释产生深远影响。特别是对于WGS ,使用更广泛的基因定义是值得的,因为变异是在全基因组范围内检测到的,并且不局限于WES 的预定义区域。GENCODE 的定期更新很好地说明了这些正在进行的改进。在过去的12 个月中,基因编码被更新了四次,最新的基因编码V38 版本,2021 年5 月更新包括超过2500 个新的蛋白质编码转录本,以及与2020 年1 月版本V33 相比的蛋白质编码基因列表中的几个修改(SUP.S2 表)。所有注释的定期更新(例如每6 个月更新一次),包括基因定义和现有样本的定期重新注释,可能会导致额外的诊断。
变异解释
除了数据分析之外,NGS 的变异解释与传统做法有很大不同,并且对分子和临床遗传学家也带来了挑战。在这里,我们描述了在临床外显子组变异解释中遇到的问题和学到的经验教训,并用实际例子加以说明。这些经验教训暂时按重要性排序,从我们经验中最有价值的经验教训开始。在所有提供的示例中,变异最初是根据我们的标准协议进行解释的,如图2 所示。我们注意到,在实践中,这些课程通常是组合使用的,我们提供的一些示例可能用于多个课程。
1. 肉眼检查数据
变异检测算法需要平衡灵敏度、特异性和性能,因此并不总能提供完美的结果。因此,肉眼检查序列比对数据(BAM/CRAM 文件)以手动过滤假阳性位点是一种很好的做法。假阳性 变异通常发生在同源性较高的区域,在检查序列比对数据时很容易看到。另一方面,变异尤其是插入 / 删除变异可能会被遗漏或不准确地检出。
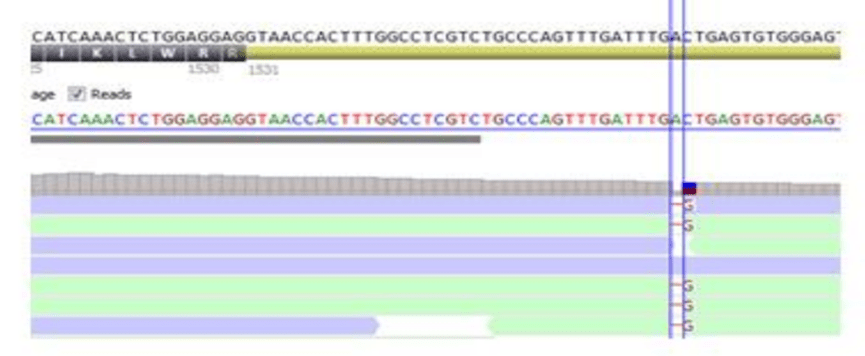

数据的肉眼检查是变异解释的一个基本方面。有几种工具可以做到这一点,包括整合基因组学查看器(IGV )。然而,对数据的目视检查非常耗时,应仅限于错误调用可能性较高的变异。这类变异包括CNV 、移码变异、等位基因比率偏离理想孟德尔比率(即不明显杂合或纯合)的变异,以及单个基因中的多个相邻变异。此外,应对实验室打算报告的所有变异进行目视检查。
2. 除了非同义单核苷酸变异以外的变异很容易被遗漏。
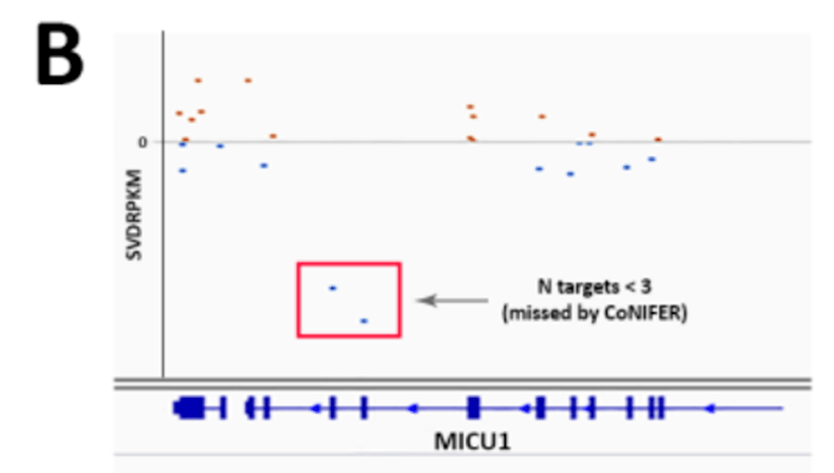
外显子组测序最初旨在检测编码区和剪接位点内的单核苷酸或多核苷酸替换,或小的缺失和重复(~1-25 bp )。近年来,多项研究表明,在一定程度上,在外显子组测序数据中也可以检测到其他类型的变异。其中包括CNV 、内含子变异、单亲二体性(UPD )、线粒体变异、重复扩增和移动元件插入。虽然与编码单核苷酸变异相比,所有这些都只能在相对较少的患者中解决病因,但这种特殊变异加在一起可以大大提高诊断率。
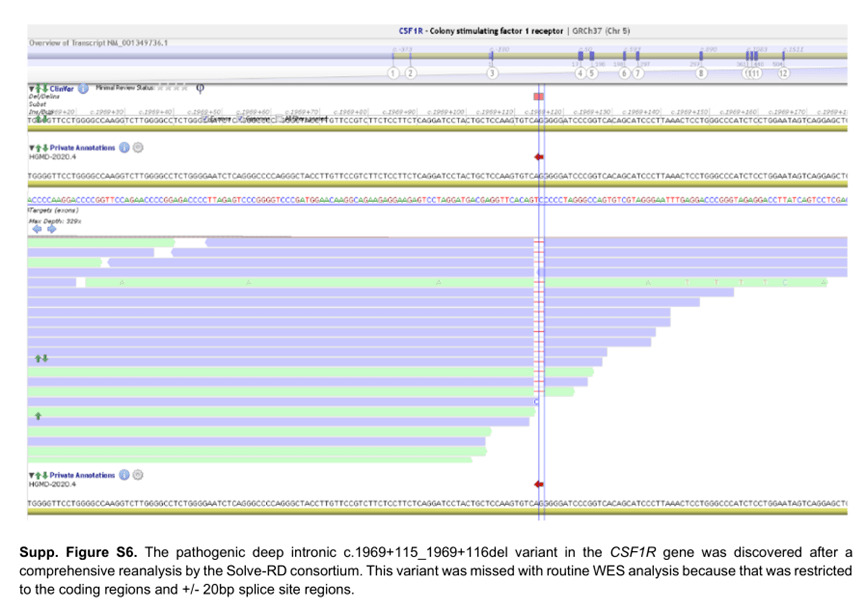
例如,对编码区和+/-20bp 剪接位点区的常规WES 分析不能诊断患有痉挛性偏瘫和关节紊乱的白质营养不良患者。作为Solve RD 联盟全面再分析的一部分,发现CSF1R 基因中的纯合已知致病性深内含子c.1969+115_1969+116del 变体(补充图S6 ),导致CSF1R 转录本中包含假外显子。虽然这个区域没有特定的捕获目标,但在这个位置,序列覆盖率足以称之为这个特定的变异。
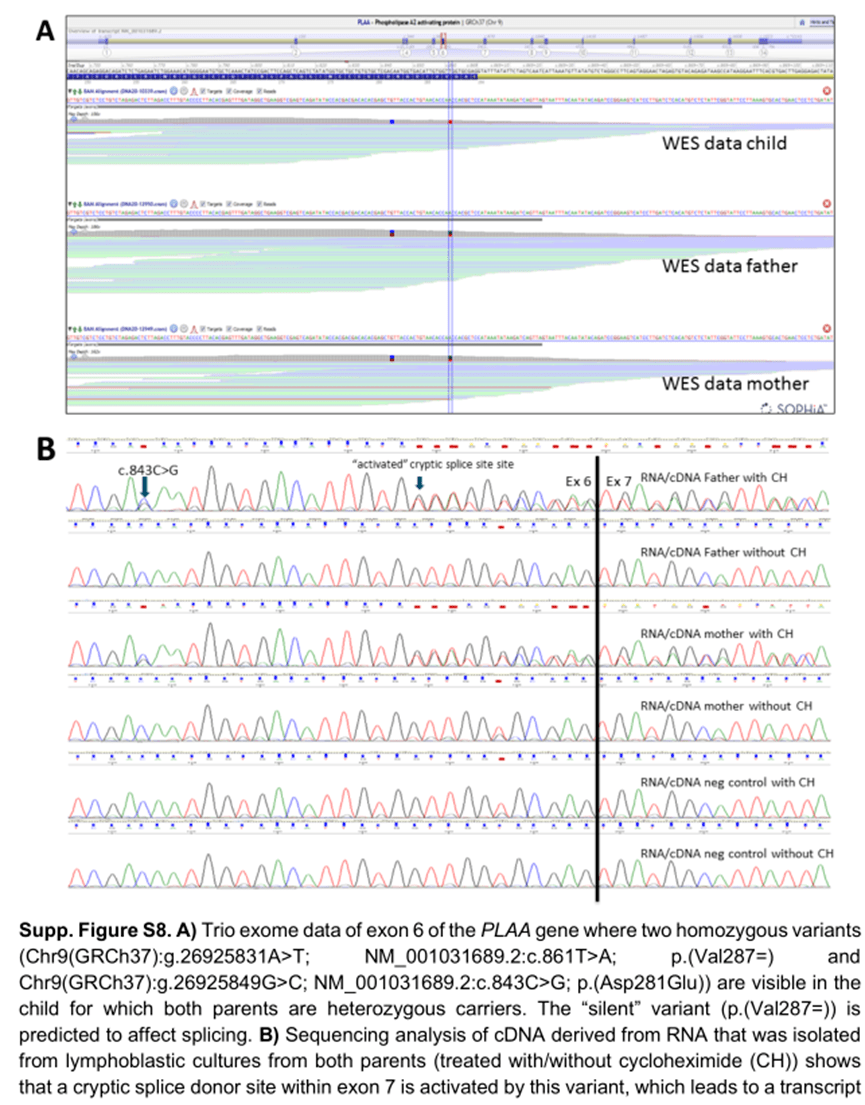
在一名患有额叶肥厚症、呼吸模式障碍和心动过速的死亡儿童中,进行了全外显子组分析。在PLAA 基因中检测到两个罕见的纯合子变异,一种错义变异和一种同义变异。虽然最初我们关注的是错义变异,但在解释后它仍然是一个VUS 。对于同义变异,剪接预测工具表明,它可能在该基因的第6 外显子中创建一个替代剪接供体位点。由于患者的临床表型符合PLAA 基因突变,因此需要对预测的剪接位点效应进行后续分析。对从携带者父母的淋巴母细胞生成的cDNA 进行测序分析,确实证实了使用了替代剪接供体位点,导致突变等位基因编码的转录本中11 个核苷酸的框外缺失(补充图S8 )。这种变异不是“ 仅仅” 是一种沉默的变异,而是导致该等位基因功能的丧失。
因此,我们建议考虑在临床上与患者表型相关的基因内的所有类型的变异,并在解释期间突出来自HGMD 和ClinVar 等数据库的所有类型的已知致病变异(即,独立于它们的位置或频率)。
3. 当其中一个“隐藏”时,复合杂合子变异很容易丢失。
另一个例子是在四名患有运动障碍的无关个体中鉴定POLR3A 基因的杂合功能缺失突变。虽然最初这些患者没有得到诊断,但经过检查,我们在所有四名患者中发现了一个额外的内含子变异(NM_007055.4:c.1909+22G>A )。这种变异的效果尚不确定,因为据预测它可以增强隐匿的供体剪接位点,同时保持原始供体剪接位点完好无损。该突变后来被证明是一种常见的亚型变异(即导致更温和的POLR3A 表型),导致19 个碱基对以组织和发育阶段特有的方式保留。
4. 还记得嵌合吗
另一个挑战是,在未受影响的父母中,致病性变异也以嵌合状态存在,在进行trio 分析时,主要关注显性基因中新发突变的检测。因此,在未受影响(嵌合)的父母身上发生的变异在孩子身上不会被标记为新发。因此,仅在寻找新发突变时,不会检测到从嵌合亲本遗传的变异。
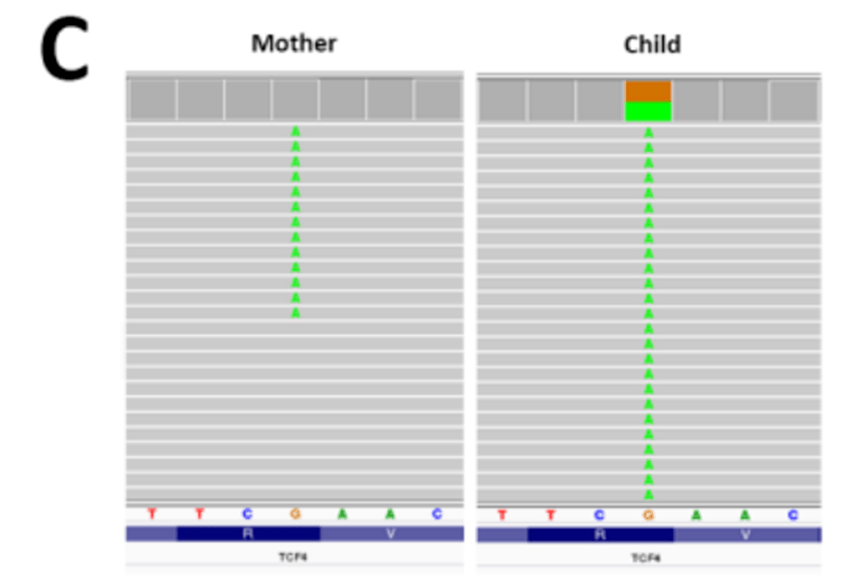
图3C
例如,我们最初错过了一个无义突变
5 染色体考虑
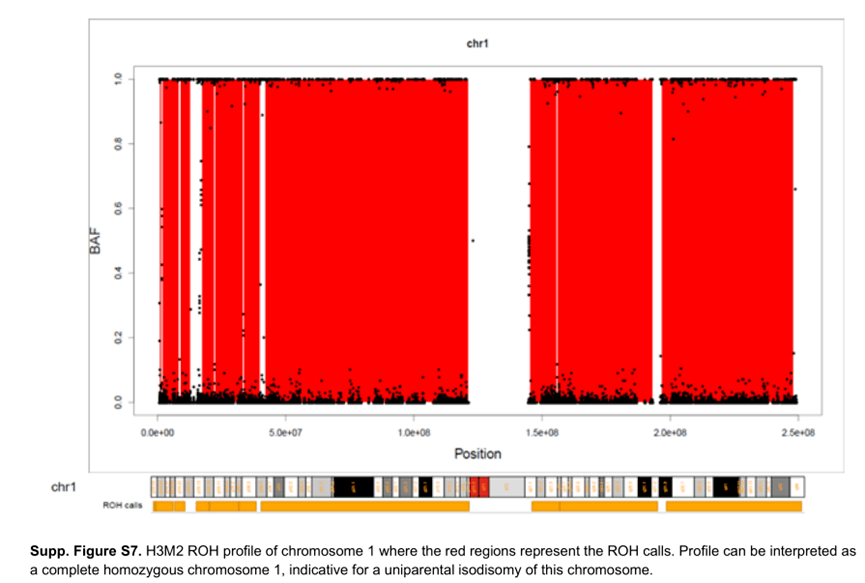
如前所述,WES 最初的目的是检测SNV (见DA-2 ),尽管可以从WES 数据分析CNV ,但在解释变异时,务必记住WES 的局限性。例如,基于测序深度算法不检测非整倍体,因为它将每个染色体的目标覆盖率标准化。我们最初错过了一例异二体X 克氏综合征(XXY ),因为当时我们实验室使用的唯一CNV 分析工具没有检测出来。因为这是两条完全相同的X 染色体,所以X 染色体上到处都是纯合性(ROH )区域,正如你在未受影响的男性身上所预期的那样。这种异二聚体X Klinefelter 是通过QF-PCR 分析,但可以通过查看WES 数据中的Y/X 覆盖率更快地检测到。
WES 中一个相对常见的拷贝数发现是检测到一条染色体上的末端重复与另一条染色体上的末端缺失相一致。这种结合是不平衡易位的明确指示,应进行常规核型分析。一个类似的事件,在一名患有严重智力残疾、发育迟缓、言语和语言缺失、肌张力减退和反流的患者中发现染色体22q13.3 上的一个约265 kb 末端缺失。因为22 号染色体是一条近端着丝粒染色体,所以在这条染色体的短臂上没有检出。同一条染色体的长臂和短臂上的这种末端缺失表明存在环状染色体。后续的核型分析显示,这确实是一个新的环状22 号染色体(补充图S9 )。区分环状染色体与“常规”末端畸变是至关重要的,因为有丝分裂期间的不稳定性是环状染色体的一个众所周知特征。
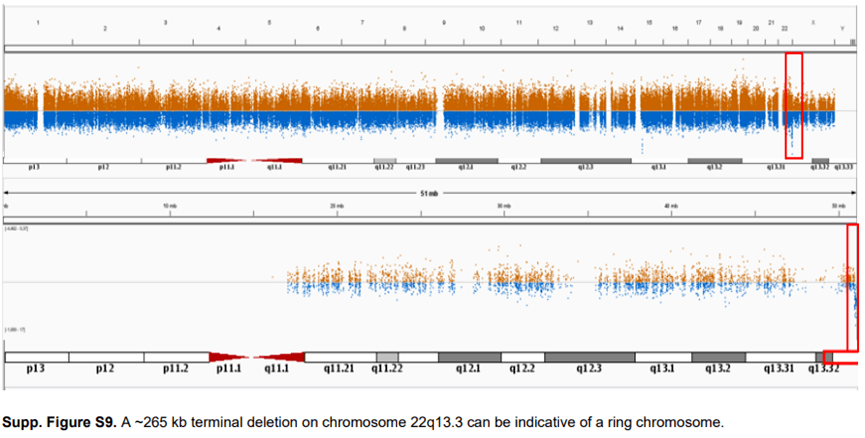
这些例子表明,也有必要具备细胞遗传学专业知识来解释WES 。从微阵列数据中解释拷贝数变异的现有指南可以为来自外显子测序数据的CNVs 的解释和后续随访提供指导。
图3D
6. 真正的致病变异可能在人群数据库中普遍存在
过滤常见变异是外显子组数据筛选的一个重要步骤,公开可用的数据库,如gnomAD ,提供来自大群体队列的聚合变异信息具有很大的帮助,这种筛选的常用阈值消除了所有等位基因频率>1% 或基于疾病频率和遗传模式的数据。当应用这种等位基因频率过滤时,有很多原因导致临床相关变异可能被错误地丢弃。
图3E
PRRT2 相关疾病的外显率估计为60% 或更高,这表明公共数据库中均聚物变化的高等位基因频率可能是由于测序误差。事实上,gnomAD 中有限的比对数据显示,在某些区域,突变等位基因的分布不均。因此,在报告之前,如果与病例相关,则必须通过另一项测试确认此类变异。虽然使用频率数据库过滤变异是一种有用的方法,但它并不完美。同样,我们建议在数据解释过程中纳入强调已知致病性变异的保障措施,以避免遗漏更高人群频率的变异(GeneRanger 软件已增加exception variant 功能,防止高频变异被遗漏)。

7. 独特的临床特征可能推动正确的诊断
数据分析有时可能会根据质量标准丢弃潜在的变异。在特定情况下,临床表型可以帮助区分变异的优先级,而无需额外的筛选步骤,甚至可以建议对特定基因进行详细分析。仅在肉眼检查测序数据后,才发现PHOX2B 基因中的新发18bp 重复事件,这是由新生儿先天性中枢性换气不足综合征的独特表型引起的。该变异未被分析出,可能是由于该区域富含GC 的重复序列中的测序读数对齐不良(图3F )。解释也是一个挑战,因为该区域在脊椎动物中并不保守(许多脊椎动物缺乏丙氨酸重复序列的重复拉伸编码),而且在gnomAD 中存在许多重叠的缺失和重复事件。然而,这种位置的重复事件是中枢性换气不足综合征的复发原因。
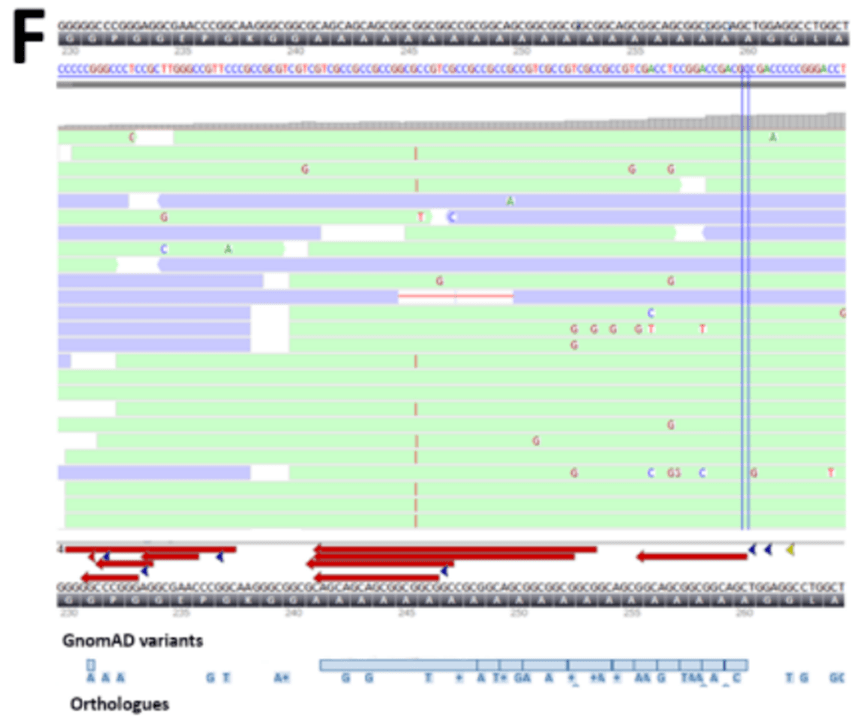
图3F
这些例子表明,患者的表型可能非常明确地指向单个基因或少量基因。不仅要注意那些可能未被call 出的基因中的变异,还要注意其他不太可能的变异,例如可能影响剪接的沉默或深度内含子变异(另见VI-2 )。因此,有专门的专家来解释特定疾病组的临床外显子组测序数据是有益的,因为这允许在他们的专业领域内更深入地了解基因病因、非典型变异类型或基因型- 表型相关性。然而,获得正确诊断的能力将始终取决于完整临床表型信息的可用性,最好是标准化格式。

8. 表型信息可能具有误导性
虽然表型信息对于正确的基因检测至关重要,但它也可能通过选择基因靶向检测阻碍基因诊断。随着基因实验室引入NGS 技术(如WES 和WGS ),转诊临床医生的诊断策略从表型优先转变为基因型优先。我们可以清楚地看到,通过或多或少的无偏测序分析,已知疾病基因中的致病性变异也可以根据基因变异的位置或类型导致非常不同的临床表型。
9. 非孟德尔遗传
WES 数据分析和解释的大多数标准过滤策略都基于经典的孟德尔遗传模式。虽然不完全外显率在遗传疾病中显然不是一种新现象,但它确实对从NGS 数据中有效过滤大量变异构成了挑战。尤其是在处理患者(健康)父母三人组数据时,变异筛选可能会导致显性基因中的遗传杂合变异被排除,或父系起源的女性或X 连锁隐性基因中杂合X 连锁变异被拒绝。
另一组具有挑战性的基因是那些具有亲本印记的基因,因此它们的表达取决于传递等位基因的亲本性别。目前已知大约有15 种由印记位点引起描述良好的疾病,但除此之外,已知或预测有数百个基因会受到基因组印记的影响( https://www.geneimprint.com/site/home) 。在一名患有多种先天性异常的患者中,我们在IGF2 基因中检测到一种新的移码变异,已知该变异存在印记,并且仅在父系等位基因上表达。由于无法从该患者的WES 数据中提取基因组定相信息,我们无法确定IGF2 变异存在于哪个等位基因上。
在这里,检测到每500-2000 个体中就有1 个发生单亲二体事件也很重要。在UPD 的情况下,两条染色体都是从同一亲本遗传的,印迹基因的变异可能是疾病的原因(上海寻因提供trio WES 分析UPD 软件)。用已知疾病机制的信息注释基因对于解释WES 数据非常有用。
10. 注意异构体、假基因和基因拷贝
长期以来,我们对基因调控的概念一直被简化为单一启动子驱动基因转录,然后剪接前mRNA ,删除所有内含子。如今,我们知道基因表达是以时间- 组织- 或发育阶段依赖的方式控制的。例如,剪接异构体可能缺少一个或多个外显子(自然外显子跳跃),具有额外的相关外显子,具有不同的翻译起始位点,或者基因可能具有多个启动子,导致不同异构体的出现。困难在于考虑哪种亚型与疾病有关,如何评估存在于仅一个子亚型中的变异,或者如果在不同亚型之间的阅读框不同,如何确保不丢失相关的“ 注释” 。
或者因为整个亚型是多余的,因此亚型特异性变异可能表现为致病性或可能是良性的。最后,由于外显子跳过,一些异构体的阅读框架部分不同,因此很难正确注释其中的变异。对于在不同亚型中具有不同效果的变异,通常可以获得所有后果,但为了方便起见,最严重的后果是优先考虑的(例如,终止密码子丢失而非错义)。然而,这可能会对一些疾病产生影响,比如努南综合征,这些疾病具有功能获得或显性负效应机制,其中错义变异是致病性的,而无义变异不是。总的来说,重要的是确保在多个异构体中调用和注释变异,然后进行正确解释,以避免遗漏相关变异。
讨论
在这里,本文提供了10 多年来从临床外显子组测序中获得的一些最重要的经验教训。作为一个诊断实验室对于质量和稳健性的关注并不鼓励持续的变化。但在这个快速发展的领域,跟上时代和创新已经成为一个必不可少的过程。通过提供我们在诊断工作流程开发过程中所犯错误的例子,我们希望我们不仅能够让人们意识到这些具体问题,而且能够让人们意识到诊断实验室中确实存在错误。对于患者和推荐临床医生了解临床外显子组测序的局限性至关重要。这些限制最好在诊断报告中提及。尽管所犯的一些错误要求我们用正确的诊断重新联系患者,但我们认为这在一定程度上是不可避免的,对犯错误的恐惧不应妨碍创新和改进,因为从长远来看,这对患者护理的危害比偶然的错误更大。
因此,重要的是要有一个全面的框架,以便在测序、数据分析和解释层面及时发现错误和问题。通过提供基准数据集,以及促进实验室之间的比较,有几项举措可以在这方面帮助实验室。从这些例子中观察到的一个有趣现象是,测序过程中出现的问题有时不是测序实验室自己发现的,而是分析数据的生物信息学家发现的。类似地,分子遗传学家在数据解释过程中经常会发现数据处理中的错误。因此,在参与临床外显子组测序过程的不同部分(即测序设施、生物信息学和数据解释)的成员之间建立常规反馈程序至关重要。
我们在这里提出的错误可能不会是我们最后的错误。从长远来看,我们努力从错误中学习,以改进诊断方法,我们希望其他人也能从我们的错误中学习。
译者介绍
边疆 男 2010年毕业于中山大学妇产科生殖内分泌专业,获博士学位。专业方向:女性生殖力保存、环境生殖毒理学。从事妇科内分泌疾病和女性生殖内分泌临床20余年
转自基因俱乐部
- 本文固定链接: https://maimengkong.com/zu/1433.html
- 转载请注明: : 萌小白 2023年4月15日 于 卖萌控的博客 发表
- 百度已收录
