千人基因组计划(1kGP)的开放获取资源主要基于来自世界五个大陆26个人群2504名个体的低覆盖全基因组测序(WGS)数据。这是第一个大型规模WGS数据集,提供了较为全面的人类遗传变异目录。
1kGP数据集包括8470万个单核苷酸变异(SNV),360万个短片段插入和删除 (INDEL),以及68818个独立的结构变异(SV)。迄今为止,1kGP资源已被引用超过18000次,并已被用于基因型(GT)估算、表达数量性状位点(eQTL)定位、变异致病性优先排序、种群历史和进化遗传学研究等基础应用。
近日,来自纽约基因组中心的研究人员与麻省总医院、耶鲁大学和人类基因组结构变异联盟(HGSVC)的研究团队合作,在 Cell 发表了题为“High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios”的文章。 该研究扩展了1kGP资源,分享了一个包含3202个样本高覆盖率WGS资源,包括602个完整的亲子三人组。研究团队通过机器学习模型集成了多种分析方法,进行了SNV和INDEL的鉴定,并生成了一组全面的SV数据集。研究团队还与前期数据集进行了比较, 强调了此次变体鉴定方法的高灵敏度和高精度,特别是对罕见SNV、INDEL和不同频率SV的检测,这些都是以前低覆盖率测序无法实现的。

文章发表在 Cell
主要研究内容
该研究队列由3202个样本组成,其中包括602个完整的亲子三人组。研究人员使用先进的方法和算法,对来自扩展队列的淋巴母细胞系衍生DNA进行测序,并达到30倍基因组覆盖的目标深度。研究团队对样本进行了SNV和INDEL检测,包括从与人类基因组参考相关的序列数据中识别变异位点,并对所有发现的变异位点进行基因分型。
研究团队共发现11717.6万个小变异位点,代表12548.4万个不同的交替等位基因,包括11104.9万个SNV和1443.5万个INDEL。在所有SNV和INDEL中,有37.5%的罕见等位基因。在所有的小变异位点中,5.9%的为新等位基因,包括6.9%的SNV和5.6%的INDEL。
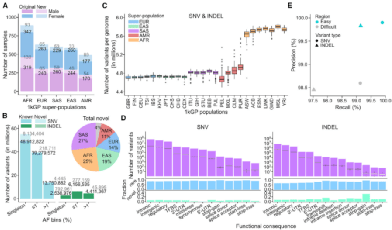
图1. 整体变体检测汇总,来源: Cell
扩展数据集中的SV
研究团队 利用3202个样本中的短读长WGS数据生成了一个SV调用集。最终的数据集合包括173366个SV,由90259个DEL、28242个DUP、673个mCNV、49693个INS、920个INV、3568个复杂SV和11个染色体间易位组成。整体SV的大小和等位基因频率分布符合预期,并观察到ALU(200-300 bp)、SVA(1-2 kb)和LINE1的移动元件特征(5 - 6kb)变体。此外,SV等位基因频率与SV大小呈负相关,且每个个体观察到的SV分布符合预期,SV调用集的精度也相当高。
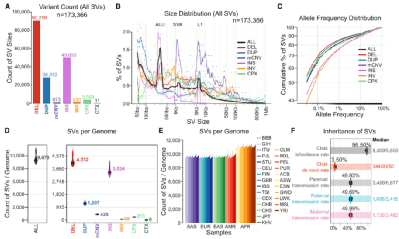
图2. 结构变异的检测和鉴定,来源: Cell
SV数据集与前期数据集的比较
接下来,研究人员将集成SV数据集和前期SV数据集进行了比较,对2504个共享样本进行分析,以评估更新后的分析流程及高覆盖率测序和基因分型能力的改进。 当前的集成SV数据集数量是前期的2倍多,并包含87.7%的前期SV数据,即在保证正确性的前提下增加了敏感性。
此外,通过与前期SV数据集比较,发现在改变基因功能的SV方面,高覆盖率的SV数据集提供了显著的附加价值,这也与前期基于短读长WGS的大种群研究结论一致。研究观察到, 每个基因组的双等位SV导致每个基因组162个基因的改变,包括97个蛋白质编码基因,50个基因的完全复制增益和15个基因的外显子重复。
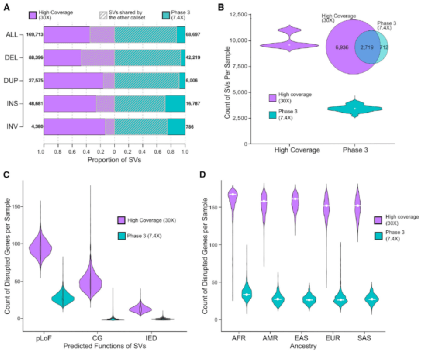
图3. SV数据集比较,来源: Cell
结 语
前期1kGP资源的一个重要作用是被用作插补参考面板,并促进了许多全基因组关联研究 (GWAS)。如今,随着原始资源的扩展,该研究团队升级了变异插补的参考数据集,包含了高覆盖率WGS和亲子三人组家族发现的更多变体。研究显示, 高覆盖WGS的变异识别(variant call)能力和精确度都有了明显提高,特别是在罕见SNV、INDEL和不同频率SV中,这些都是以前低覆盖率测序无法实现的。
“千人基因组计划队列是宝贵的资源,我们认为使用最新版本的短读长测序技术更新测序数据,同时增加先前遗漏的家庭样本丰富性,这对整个研究领域很有用。” 文章共同通讯作者、纽约基因组中心计算生物科学主任Michael Zody教授解释说。
文章共同第一作者、纽约基因组中心高级生物信息学科学家Marta Byrska-Bishop博士 表示:“新的变异插补包括更多位点,尤其是更多常见的INDEL和SV,从而扩大了GWAS可利用的变体数量,鉴于非SNV变异的影响大小,很可能能够发现新的遗传关联,帮助查明致病变异。”
这些数据已经引起了遗传学和基因组学界的兴趣。据悉,所有原始序列数据和变异调用集在测序完成后立即通过几个基因组数据存储库向公众发布,包括由欧洲分子生物学实验室欧洲生物信息学研究所共同维护的国际基因组样本资源(IGSR)(EMBL-EBI)。
“我们的目标是让这一公共资源成为未来人口遗传学研究和方法开发的基准。” 文共同第一作者、麻省总医院基因组医学中心博士后Xuefang Zhao博士补充道。
参考文献:
1. Marta Byrska-Bishop et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell, 2022.
2. Abel, H.J., et al. (2020). Mapping and characterization of structural variation in 17, 795 human genomes. Nature.
3. Ebler, J., et al. (2022). Pangenome-based genome inference allows efficient and accurate genotyping across a wide spectrum of variant classes. Nat. Genet. 54, 518–525.
·END ·
- 本文固定链接: https://maimengkong.com/zu/1431.html
- 转载请注明: : 萌小白 2023年4月14日 于 卖萌控的博客 发表
- 百度已收录
