终于到了最重要的关联分析了。
我们先回顾一下我们前几节课都讲了什么,第一节告诉您如何安装软件,第二节告诉您应该如何处理表型,第三节,vcf文件的处理,第四节,群体结构分析,第五节,亲缘关系分析。
那这节我将教大家如何去运用前几节讲的东西,去做GWAS。进行分析之前所需要准备的材料有:TASSEL软件、群体结构Q矩阵文件、亲缘关系Kinship文件、基因型文件、表型文件。
我们先ls-t一下,4个文件已经准备齐全:

我们在亲缘关系那节讲过,tassel的第一步就是给vcf文件进行排序,排成人家认可的顺序,接下来我们先运行GLM模型看一下,命令行如下:
run_pipeline.pl -fork1 -vcflecture09.genotype.vcf -fork2 -r lecture09.trait.txt -fork3 -q lecture09.Q.txt-excludeLastTrait -combine4 -input1 -input2 -input3 -intersect -glm -exportlecture09_glm_ -runfork1 -runfork2 -runfork3
运行结束之后我们ls-t看一下:
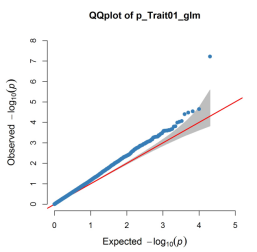
生成了2个文件,我们less -S lecture09_glm_1.txt这个文件看一下:
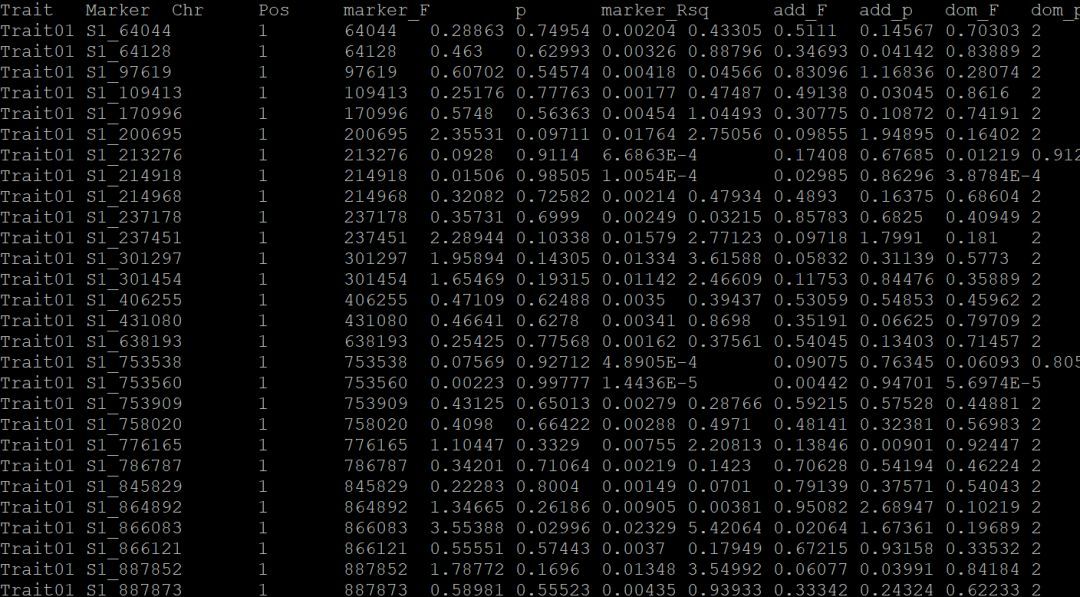
第一列是你的性状名字,第二列是标记,第三列是染色体,第四列是染色体位置。。。
然后根据你所设置的阈值,你就可以把你想要的显著位点筛选出来啦!
MLM模型的运算时间过长,我就不演示啦,命令行加了个Kinship矩阵:
perl tassel_v5/run_pipeline.pl-fork1 -h lecture09.hmp.txt -fork2 -r lecture09.trait.txt -fork3 -qlecture09.Q.txt -excludeLastTrait -fork4 -k lecture09.K.txt -combine5 -input1-input2 -input3 -intersect -combine6 -input5 -input4 -mlm -mlmVarCompEst P3D-mlmCompressionLevel None -export lecture09_mlm_ -runfork1 -runfork2 -runfork3-runfork4
当然能进行全基因组关联分析的软件有很多,我们在这里只给大家讲了其中的一个经典软件,模型呢,讲了2个经典的GLM、MLM,不同的性状,适合不同的模型,找到适合您的模型才是最好的,如果您想学习更多的方法,可观看基迪奥的GWAS系列课程。
课程中还讲解了EMMAX,FastLMM、GAPIT等一系列方法,同时也会教大家如何快速的大批量筛选显著位点,同时也会教您如何用关联分析结果绘制发表级别的图片。
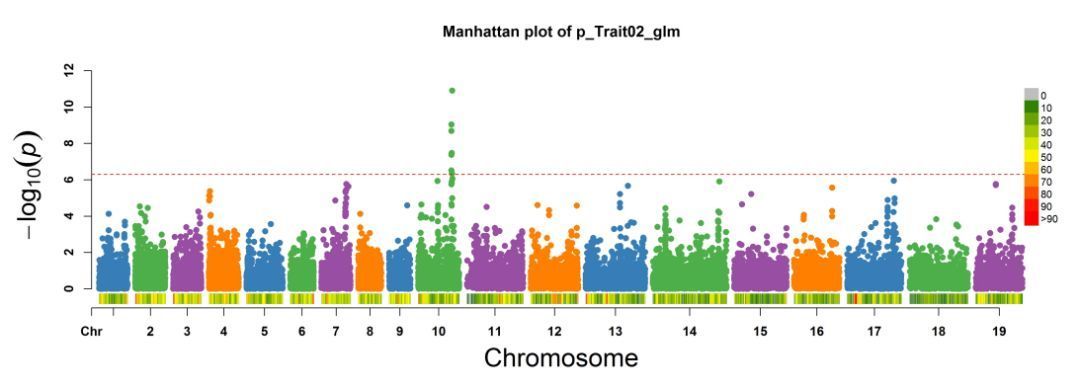
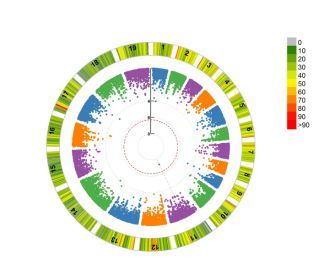
GWAS系列到这里就结束了,期待我们在课程中相遇,您可以加QQ群:(154447756)与我们一起探讨。
教程链接:http://www.omicshare.com/class/
- 本文固定链接: https://maimengkong.com/zu/1426.html
- 转载请注明: : 萌小白 2023年4月13日 于 卖萌控的博客 发表
- 百度已收录
