表观遗传学领域的研究是近年来生命科学领域的热点之一,高通量测序技术的进步提供了各种破译表观遗传学图谱的方法,其中包括研究染色质可及性的ATAC-seq。2013年Buenrostro J.D.等[1]发表ATAC-seq(Assay for Transposase-Accessible Chromatin with highthroughput sequencing)技术,由于起始细胞数要求低(大约50,000个细胞)、操作简单和实验周期短等优势而得到广泛应用。但是,ATAC-seq的生信分析流程和软件却还没有统一标准。
很多ChIP-seq分析软件被用于ATAC-seq分析,但是这些软件在ATAC-seq分析过程中是否完全适用还存在疑问,我们就来看一下澳大利亚莫纳什大学的Yan F.等人于2020年2月在Genome Biology上发表的综述《From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis》是怎么说的吧。

ATAC-seq技术背景
哺乳动物的DNA通过三个主要的层次尺度进行高度压缩:
a. 第一层是核小体,DNA缠绕在核小体单体上;
b. 第二层是染色质,核小体单体互相缠绕形成染色质;
c. 第三层是染色体,染色质进一步压缩为染色体。
DNA压缩的三个尺度及其相互作用共同造就了基因表达调控。
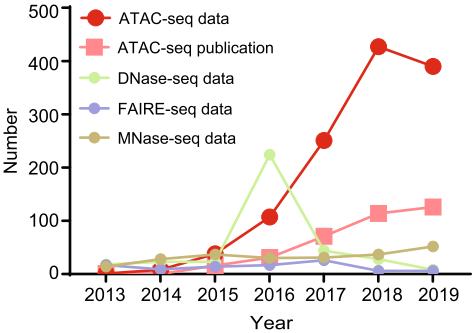
ATAC-seq技术自从发表以来,从众多方法中脱颖而出,成为研究染色质可及性的主要手段。近年来,ATAC-seq测序数据上传数量和文章发表数量呈指数增长,体现了该技术在生物学问题中的价值。
ATAC-seq实验使用基因工程改造的超高活性Tn5转座酶(Hyperactive Transposase Tn5),该Tn5酶已经预载了测序文库构建时的Adaptor(P5和P7)。Tn5酶可以切割开放的染色质区域,在酶切位点留下9 bp的粘性末端,并将两种Adaptor连接到切口的粘性末端上;随后DNA片段会被扩增形成文库被测序。
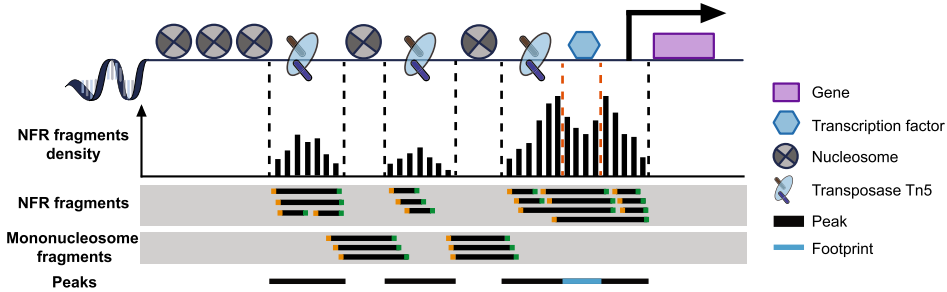
下图比较直观地展示了ATAC-seq的技术原理,其中NFR fragments是开放染色质中两个核小体之间的Linker DNA片段,由Peak Calling分析得到的Peak来鉴定;蓝色的Footprint则是转录因子的足迹,对应的是转录因子足迹分析;核小体单体(Mononucleosome)上结合的DNA片段则反映出核小体的位置信息,对应的是核小体占位分析。
尽管ATAC-seq实验方法相对简单且稳定,但是专门为ATAC-seq测序数据开发的生物信息学分析软件却非常少。由于ATAC-seq和ChIP-seq数据的相似性较高,ChIP-seq分析使用的软件一般也可用于ATAC-seq的分析,但是使用ChIP-seq软件分析得到的ATAC-seq结果尚未得到系统性的评估。因此这篇综述针对用于ATAC-seq分析的现有软件资源做出全面性的介绍和评估。
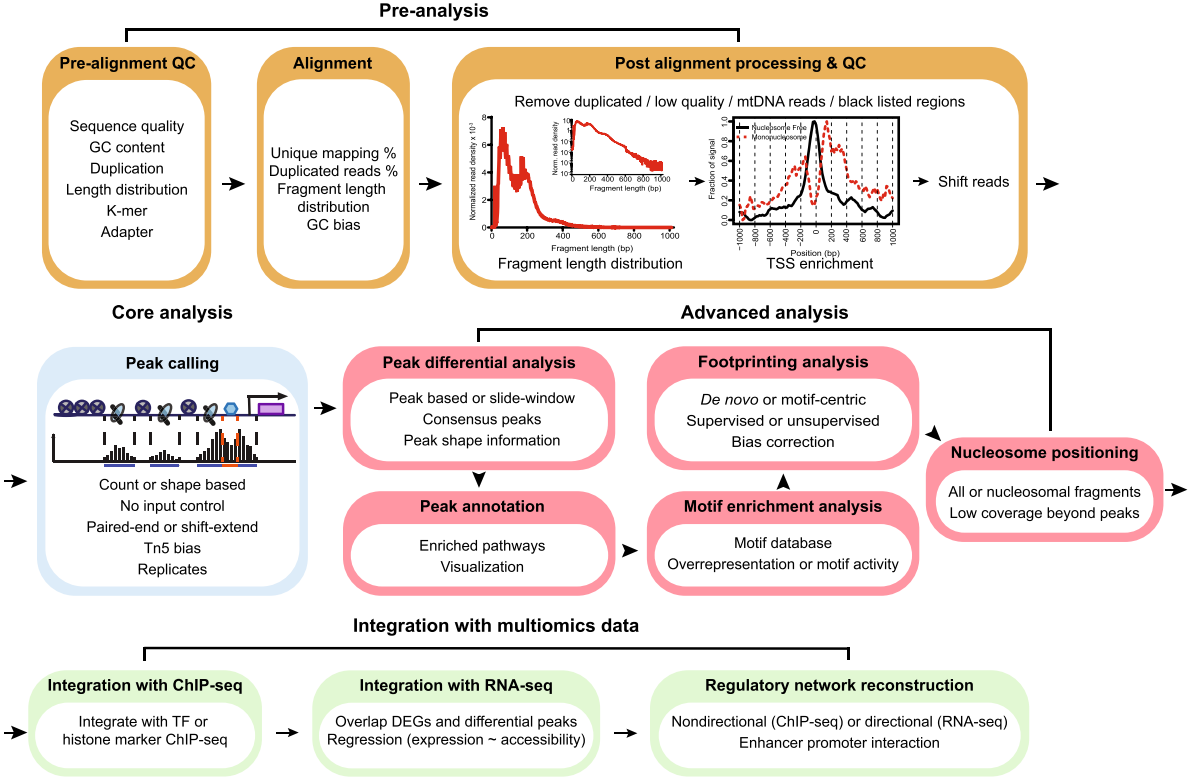
ATAC-seq分析流程
ATAC-seq预分析
ATAC-seq分析的第一步是预分析,主要包括三个部分:1. 测序原始数据质控;2. 序列比对(Mapping);3. 比对后处理和质控。
一、测序原始数据质控
对ATAC-seq的测序原始数据质控和序列比对的流程与其它二代测序数据标准分析流程基本相同,比如可以选择FastQC软件来可视化碱基质量得分、GC含量、序列长度分布、序列重复水平、k-mer比例是否过高以及引物二聚体和接头自连情况等。ATAC-seq文库一般采用的是Illumina Nextera建库方法,接头和Truseq文库不一样,所以使用去除接头序列软件cutadapt、AdapterRemoval v2、Skewer或trimmomatic时需要提供Nextera文库的接头序列。
二、序列比对
去除低质量碱基和接头序列后,可以再用FastQC软件做一下质控。质控合格的reads就可以比对(Mapping)到参考基因组上。BWA-MEM和Bowtie2这两种软件用于双端短序列比对上更快,这两个软件中的soft-clip策略保留了突出和没有比对上的碱基,可以增加在参考序列上有唯一比对位置的序列数目(Unique比对率)。对于哺乳动物来说,Unique比对率大于80%的数据是比较成功的ATAC-seq文库。
三、比对后处理和质控
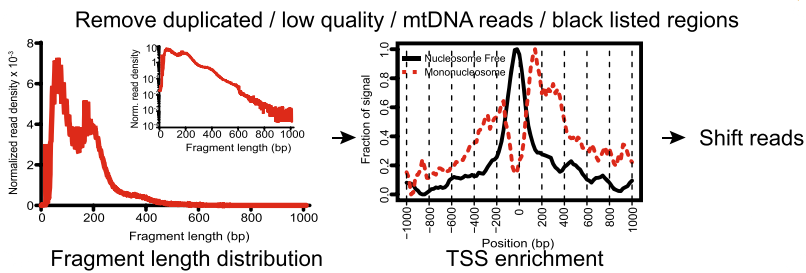
序列比对后可以得到BAM后缀的文件,可以用Picard和SAMtools软件来统计Unique比对率、重复reads比例和片段大小分布。Mapping后需要去除对后续分析产生干扰的reads:1. 配对错误和比对质量比较低的reads需要被剔除;2. 线粒体基因组由于没有染色质组装,处于开放状态,更容易被Tn5酶切割,所以线粒体序列需要去除;3. ENCODE数据库中黑名单区域(blacklisted regions)包含了一些异常、read覆盖度很高的区域,在这些区域的reads需要去除;4. 由PCR建库过程产生、重复率过高的reads也需要去除。
除此之外,测序文库插入片段大小的分布也可以用来判断ATAC-seq实验的质量。插入片段大小的理论分布为:NFR fragments(<100 bp)、核小体单体(~200 bp)、核小体二聚体(~400 bp)和核小体三聚体(~600 bp),每个位置上都会有对应的特征性的峰分布(见下图左)。NFR fragments应该富集在转录起始位点(TSS)附近(见下图右黑色实线),而结合核小体的区域在TSS位置应该缺失且在TSS两侧相对富集(见下图右红色虚线)。
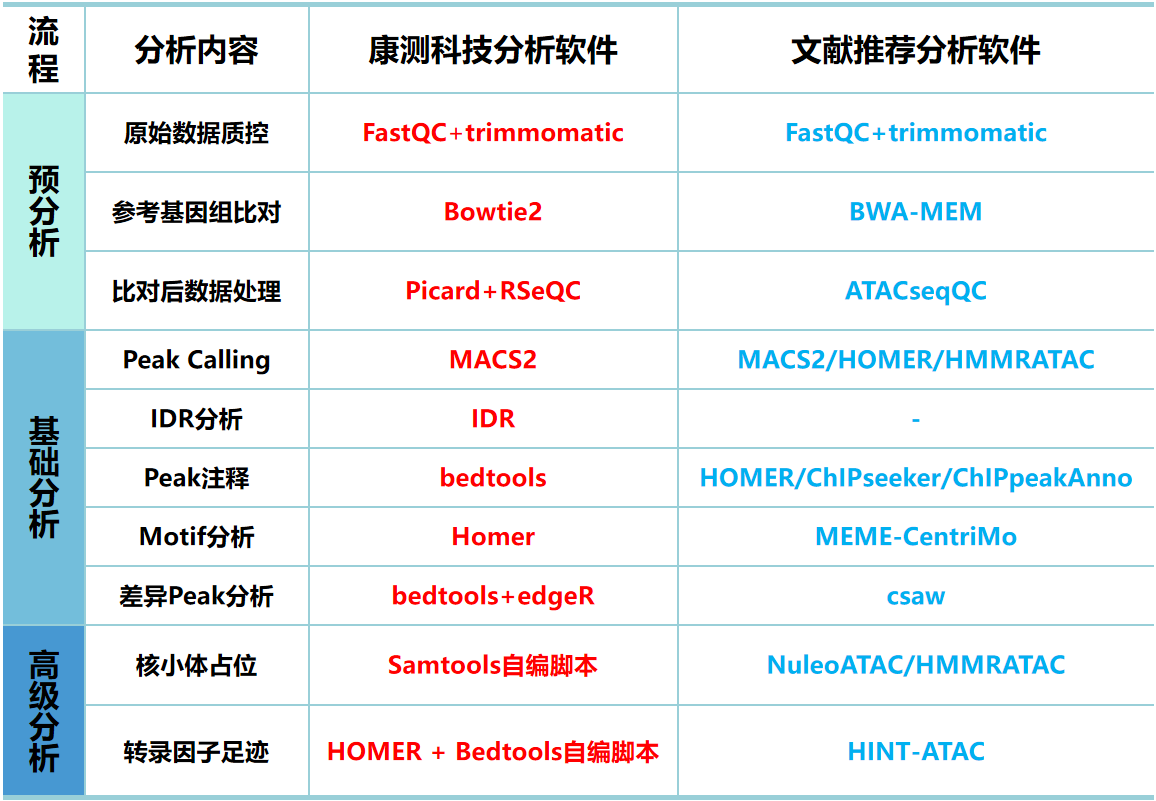
文章推荐软件组合:FastQC → trimmomatic → BWA-MEM → ATACseqQC
康测科技软件组合:FastQC → trimmomatic → Bowtie2 → Picard+RSeQC
ATAC-seq核心分析:Peak Calling
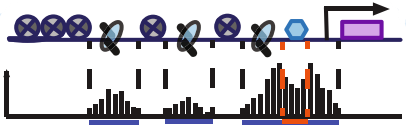
ATAC-seq分析的第二步是识别染色质开放区域,即Peak Calling。许多分析ChIP-seq数据的Peak Calling软件可用于ATAC-seq数据,而ENCODE选择MACS2作为ATAC-seq的标准Peak Calling软件。与ChIP-seq不同的是,由于Tn5酶切割的随机性和成本原因,ATAC-seq没有Input数据作为对照,所以需要Input数据的Peak Calling软件不能用于分析ATAC-seq数据。ATAC-seq数据中包含了NFR reads和DNA与核小体结合区域的reads,而ATAC-seq主要关注NFR部分的reads,所以不能直接用所有reads进行Peak Calling。
一种方式是把NFR reads单独提取出来进行分析;另一种方式是采用shift-extend的方法进行分析,这种方法尝试对Tn5酶切口的末端平滑化事件进行计数(见下图)。第二种方法更为通用,因为这种方法几乎适用所有为ChIP-seq数据开发的Peak Calling软件,并且不受插入片段大小的影响。
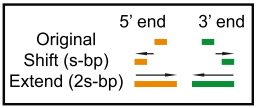
ChIP-seq Peak Calling软件根据原理主要分为两大类:Count-based方法和Shaped-based方法。一般Count-based方法的软件更易于使用和解释结果。这些软件采用不同的统计方法比较目标区域和随机背景区域的reads分布形状,常用的软件包括:
a. 假设片段分布为泊松分布:MACS2、HOMER、SICER/epic2
b. 假设片段分布为零膨胀负二项分布:ZINBA
c. 核密度估计来判断片段分布:F-seq、PeakDEck
d. 不使用片段分布假设但通过软件打分:SPP
e. 混合模型:JAMM
其中F-seq和ZINBA软件更新维护不及时,使用的时候应该注意。
Shaped-based方法直接或者间接利用reads的密度分布信息进行Peak Calling,包括PICS、PolyaPeaK和CLC等软件,但这些软件暂时还没有用于ATAC-seq数据的分析。
目前专门为ATAC-seq开发的Peak Calling软件只有HMMRATAC。该软件通过三状态半监督隐马尔科夫模型算法把基因组分成高信号强度的活性染色质区域、中等信号强度的核小体区域和低信号强度的背景区域。虽然HMMRATAC计算量偏大,耗时较长,但其结果表现比MACS2和F-seq更好,还可以同时提供核小体的位置信息。
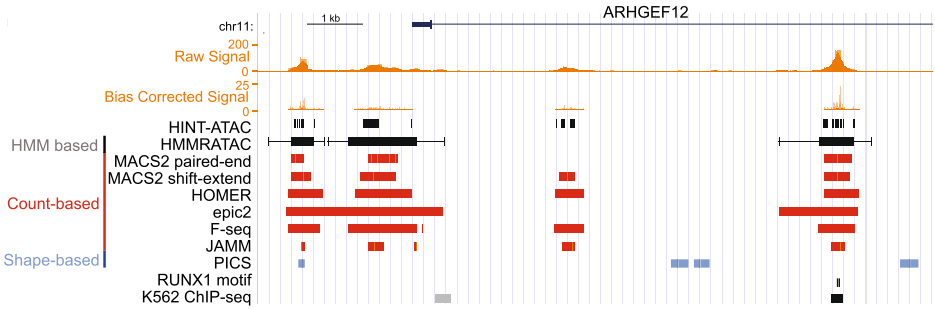
通过上述软件分析得到的Peak可视化情况如上图。Count-based方法的软件结果表现差异不大,但Shaped-based方法的软件结果与Count-based软件结果非常不同。
目前仍没有综合性指标来评估这些Peak Calling软件的结果表现。
以上分析了各个Peak Calling软件的结果表现,但是并没有针对存在生物学重复设置的Peak Calling结果可信度进行探讨。康测科技引入IDR分析来判断存在生物学重复时Peak Calling结果的可信度(见下图)。IDR(Irreproducibility Discovery Rate)是指不可重现的发现率,用于测量生物学重复中的可重现性。ATAC-seq分析中是通过比较一对经过排序的regions/peaks的列表,然后计算反映其重复性的值。因此通过IDR分析结果得到的Peak即是可信度更高的Peak。康测建议每组样品设置2个及以上的生物学重复。
文章推荐Peak Calling软件:MACS2/HOMER/HMMRATAC
康测科技Peak Calling软件:MACS2 (shift-extend method) + IDR
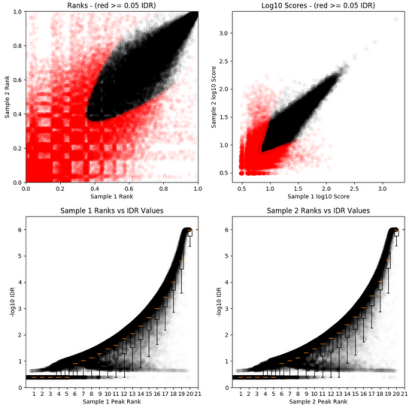
IDR分析结果展示
ATAC-seq高级分析
ATAC-seq的主要功能是揭示转录调控的各个方面,其第三步分析要在4种水平对结果进行分析和解释:1. Peak注释和差异Peak分析;2. Motif分析;3. 核小体占位分析;4. 转录因子足迹分析。
一、Peak注释和差异Peak分析
一般情况下,软件会关联Peak与其最近的基因或者调控元件来进行Peak注释,HOMER、ChIPseeker和ChIPpeakAnno这三个软件都可以把Peak分配到最近或重叠的基因、外显子、内含子、启动子、5’UTR、3’UTR和其它基因组功能区。随后可以用Gene Ontology(GO)、KEGG和Reactome等数据库做Peak关联基因功能富集分析。ChIPseeker和ChIPpeakAnno软件都具有可视化功能。
目前还没有专门为ATAC-seq开发的差异Peak分析软件。差异Peak分析首先通过寻找候选区域(共有Peak或根据bin划分的基因组),然后进行标准化,再对落在这些区域里的片段进行计数,最后在相同坐标内与其它处理条件的样本进行统计学比较。在以共有Peak为基础分析的软件中,HOMER、DBChIP和DiffBind依赖RNA-seq差异表达基因分析中使用的R包计算差异Peak,例如edgeR、DESeq和DESeq2等,所以这些软件分析时都要求设置生物学重复。
HOMER把所有生物学重复样品的数据合并到一起以减少差异peak的假阳性结果。DBChIP和DiffBind通过取交集或并集的方法得到共有Peak,不过取交集的方法有时会忽略一些样本或特殊的Peak,而取并集则会使假阳性结果增多。另外一些不依赖RNA-seq分析R包的软件包括PePr、DiffReps和ChIPDiff,还有一种edgeR包的扩展软件csaw,这些软件使用滑窗(Sliding window)的方法进行分析,但是得到的结果假阳性率很高,需要设置比较严格的FDR。
文章推荐Peak注释与差异分析软件:HOMER/ChIPseeker/ChIPpeakAnno + csaw
康测科技Peak注释与差异分析软件:Bedtools + edgeR
二、Motif分析
开放的染色质区域一般可以结合特定的转录因子进而影响转录过程,转录因子结合识别的DNA序列即为motif,人体中大约有1600种转录因子,其中一半多已经有明确报道的motif。对motif的分析包括motif富集分析和转录因子Footprint(足迹)分析。
-
Motif富集分析
目前有很多motif数据库,其中使用最普遍的是JASPAR数据库,该数据库收录了很多物种的motif数据,可以通过APIs或者Bioconductor的R包下载相关数据。除此之外,CIS-BP和TRANSFAC数据库收录了真核生物转录因子的motif信息,HOCOMOCO数据库则专门收录了人和小鼠的motif,RegulonDB为大肠杆菌的motif数据库。
Motif富集分析过程如下:首先获得每个Peak区域里motif的位置和频率,然后与随机背景或另一种条件的背景进行比较,最后得到motif富集的结果。HOMER、MEME-AME、MEME-CentriMo和DAStk分别采用不同的统计检验的方法来比较Peak和背景区域的motif出现的频率差异。
文章推荐Motif富集分析软件:MEME-CentriMo
康测科技Motif富集分析软件:HOMER
-
转录因子足迹分析
另一种通过ATAC-seq解释转录因子调控方式的是足迹分析。转录因子的足迹是指一个转录因子结合在DNA上,阻止Tn5酶切割,在染色质开放区域留下一个相对缺失的位置。然而,做足迹分析有三个问题需要解决:1)由于建库时Tn5酶切时会产生9 bp的粘性末端切口,经过末端修复补齐后,原始reads在预处理时需要经过移位才可以准确检测到Footprint;2)Tn5酶切具有5’端偏好性;3)某些瞬时结合的转录因子足迹信号比较弱。
足迹分析软件根据算法可以分为两大类:de novo和Motif-centric。de novo类型的软件需要通过理论计算来鉴别转录因子的足迹信息,并且消除Tn5酶切时的5’偏好性。目前只有HINT-ATAC可以处理ATAC-seq数据特有的偏好性。
Motif-centric方法主要关注已知TF的结合位点,主要软件有MILLIPEDE、DeFCoM等。联合ChIP-seq数据的Motif-centric方法在足迹分析上优于de nove的方法,但是这些ChIP-seq数据来源于特定的转录因子和特定的细胞类型,通用性并不强。而de novo的方法在一些低质量和新发现的一些motif上具有优势。
文章推荐转录因子足迹分析软件:HINT-ATAC
康测科技转录因子足迹分析软件:HOMER + Bedtools自编脚本
三、核小体占位分析
核小体单体可以结合大约147 bp的DNA,在标准的ATAC-seq文库中,较长的插入片段对应DNA与核小体结合的区域。ATAC-seq数据中核小体结合区域比染色质开放区域reads覆盖度更低,所以相比MNase-seq,ATAC-seq的核小体占位分析难度更高。在一般情况下,为MNase-seq开发的软件(比如DNAPOS2、PuFFIN、iNPS和NucTools)可用于ATAC-seq。专门为ATAC-seq开发的软件包括NuleoATAC和HMMRATAC。NuleoATAC比DANPOS2结果表现更好,而HMMRATAC可以同时完成Peak Calling和核小体占位分析。
文章推荐核小体占位分析软件:NuleoATAC/HMMRATAC
康测科技核小体占位分析软件:Samtools自编脚本
ATAC-seq与多组学数据联合分析
1. 转录因子ChIP-seq:由于大部分转录因子结合的是染色质开放区域,所以ATAC-seq的Peak可能和转录因子ChIP-seq的Peak存在部分重叠的情况,而且ATAC-seq得到的Peak长度往往更长,因此ATAC-seq数据和转录因子ChIP-seq数据可以相互验证。转录因子在ChIP-seq中独有的Peak暗示这个转录因子可能是结合在异染色质区域的驱动型转录因子(Pioneer TFs),驱动型转录因子随后招募染色质重塑复合体以及其它转录因子开始转录。另外,联合分析已经报道的ChIP-seq数据可以更准确地分析转录因子的足迹。
2. 组蛋白修饰ChIP-seq:ATAC-seq数据同样可以和组蛋白修饰ChIP-seq数据进行联合分析,其中转录激活性修饰(H3K4me3,H3K4me1和H3K27ac等)与染色质开放程度呈正相关,转录抑制性修饰(H3K27me3)与染色质开放程度呈负相关。联合已知的增强子和启动子之间的相互作用数据也可以帮助构建调控网络。
3. RNA-seq:ATAC-seq数据可以通过联合分析RNA-seq数据来发现哪些差异表达的基因是受染色质可及性调控的,进一步可以推测这些差异表达的基因哪些是受开放染色质中具有motif和footprint的转录因子调控的,因此ATAC-seq与RNA-seq的联合分析有助于破译基因调控网络和细胞异质性。
总结
ATAC-seq近年来发展迅速,已成为研究染色质可及性的主流方法。该review系统性地描述了ATAC-seq生信分析的主要流程,并推荐了相关软件:使用FastQC进行质控;trimmomatic去除低质量碱基和接头序列;使用BWA-MEM作为序列比对软件;使用MACS2进行Peak Calling;使用csaw进行差异Peak分析;使用MEME-CentriMo寻找motif以及富集分析;使用ChIPseeker用来进行Peak注释和可视化;使用HMMR-ATAC来分析核小体占位;使用HINT-ATAC进行转录因子的Footprint分析。
康测科技与这篇综述推荐的ATAC-seq分析流程相比稍有不同:在Peak Calling步骤引入IDR分析来判断Peak可信度;差异Peak分析使用bedtools和单独的edgeR包结合自编脚本提高差异Peak分析中参数设置自由度等,本质都是为了得到准确度和可信度更高的分析结果。
该review对于大家理解ATAC-seq分析的流程,解释ATAC-seq分析结果有着非常大的帮助,不失为一篇参考价值极高的文章。
分析软件对比汇总
参考文献
[1] Buenrostro, J.D., et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods, 2013, 10(12): p. 1213-8.
转自:www.seqhealth.cn/view/158.html- 本文固定链接: https://maimengkong.com/zu/1338.html
- 转载请注明: : 萌小白 2023年1月13日 于 卖萌控的博客 发表
- 百度已收录
