学习章节
https://bioconductor.github.io/BiocWorkshops/r-and-bioconductor-for-everyone-an-introduction.html#introduction-to-bioconductor
文章目录
- 学习章节
- 1. Bioconductor的一些补充小用法
- 2. Working with Genomic Ranges
- 2.1 importing data 导入bed文件
- 2.2 Working with genomic ranges
- 2.3 Genomic annotations
- 2.4 Overlaps
1.Bioconductor的一些补充小用法
使用valid()查看包安装的版本情况
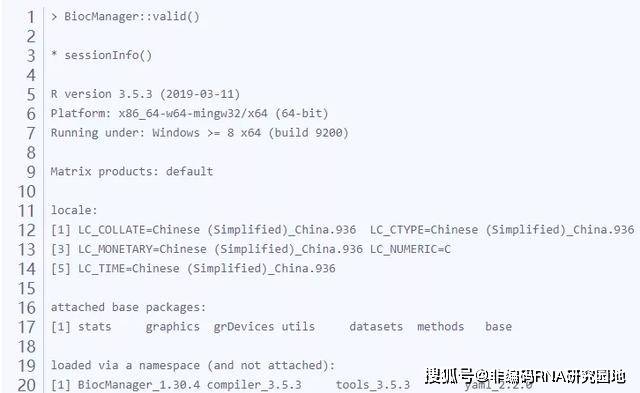
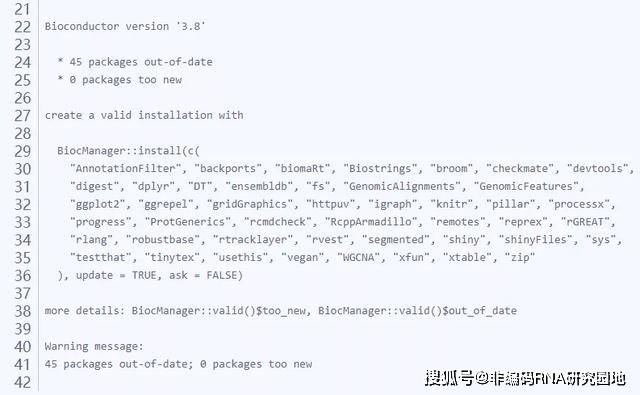
avaliable() 用于搜索相关的包
1 ## 例如这里输入TxDb.Hsapiens,它会自动匹配相关的包
2 BiocManager::available("TxDb.Hsapiens")
3 #> [1] "TxDb.Hsapiens.BioMart.igis"
4 #> [2] "TxDb.Hsapiens.UCSC.hg18.knownGene"
5 #> [3] "TxDb.Hsapiens.UCSC.hg19.knownGene"
6 #> [4] "TxDb.Hsapiens.UCSC.hg19.lincRNAsTranscripts"
7 #> [5] "TxDb.Hsapiens.UCSC.hg38.knownGene"
通过网站https://support.bioconductor.org/ 来查询解决和包相关的问题
2. Working with Genomic Ranges
- · Importing data
- · Working with genomic ranges
- · Genomic annotations
- · Overlaps
rtracklayer包提供函数import() 帮助用户读取基因组格式的文件(例如:BED,GTF,VCF,FASTA) 并封装成Bioconductor下的对象。GenomicRanges包提供了多种函数,来在基因组坐标系下操纵各种数据。
2.1 importing data 导入bed文件
1 library(rtracklayer)
2 ## 使用file.choose() 来选择文件
3 fname <- file.choose() # CpGislands.Hsapiens.hg38.UCSC.bed
4 fname
5 file.exists(fname)
6 ## 使用import()函数导入bed文件。导入后,将以GenomicRanges的对象描述这份CpG岛数据。
7 cpg <- import(fname)
8 cpg
注意 BED格式的文件,它们的坐标体系是0-based的,并且intervals是半开区间(start在范围内,end在范围后一个坐标里)
而Bioconductor得坐标是1-based,并且是闭区间(start和end坐标都包含在范围内),因此使用import()函数导入数据得时候,它会自动将bed文件坐标转换为Bioconductor文件对象的坐标。
2.2 Working with genomic ranges
1 ## 使用函数 keepStandardChromosomes 保留标准染色体,标准染色体指的是chr1-22和x,y染色体
2 ## 很多时候我们获取的数据里 可能染色体不止chr1-22与x,y ;
3 ## 可能还包含其它染色体,例如:chr22_KI270738v1_random这样的染色体。
4 ## 通过keepStandardChromosomes 就可以去除这些其它的染色体,只保留标准染色体
5 cpg <- keepStandardChromosomes(cpg, pruning.mode = "coarse")
6 cpg
GenomicRanges对象包含两个部分
必需的:seqnames(染色体号),start(起始位点),end(终止位点),strand (正链或负链)
非必需的:另外的元素,例如本例中的name。
必需的元素内容,可以使用函数start(), end(), width()获取。非必需的元素,使用$符号获取内容。
1 head( start(cpg) )
2 #> [1] 155188537 2226774 36306230 47708823 53737730 144179072
3 head( cpg$name )
4
5 ## 使用subset()函数获取染色体1号和2号上的cpg岛
6 subset(cpg, seqnames %in% c("chr1", "chr2"))
2.3 Genomic annotations
1 ## 导入注释数据
2 library("TxDb.Hsapiens.UCSC.hg38.knownGene")
3 ## 查看注释数据中的所有转录本
4 tx <- transcripts(TxDb.Hsapiens.UCSC.hg38.knownGene)
5 tx
6 ## 为了演示方便,目前只保留标准染色体
7 tx <- keepStandardChromosomes(tx, pruning.mode="coarse")
8 tx
2.4 Overlaps
可以使用findOverlaps(),nearest(),precede()和follow()函数,来完成overlaps。
1 ## count the number of CpG islands that overlap each transcript
2 olaps <- countOverlaps(tx, cpg)
3 length(olaps) # 1 count per transcript
4 #> [1] 182435
5 table(olaps)
6 ## 将计算的overlaps加入GR对象
7 tx$cpgOverlaps <- countOverlaps(tx, cpg)
8 tx
9 ## subsetting the GRanges objects for transcripts satisfying particular conditions, in a coordinated fashion ## where the software does all the book-keeping to makes sure the correct ranges are selected.
10 subset(tx, cpgOverlaps > 10)
文章来源:Yujia_compbio
- 本文固定链接: https://maimengkong.com/zu/1312.html
- 转载请注明: : 萌小白 2022年12月16日 于 卖萌控的博客 发表
- 百度已收录
