ChIP-seq试验普遍用于探究蛋白质与核酸的结合作用。例如最常见的,期望获得某个转录因子的功能,查看它结合到哪些基因启动子区域并调控它们的转录,以及更高级的分析识别该转录因子的motif位点等,就可通过ChIP-seq来实现。
作了ChIP-seq试验,经过初步下机处理并比对参考基因组之后,获得了目标蛋白在基因组区域的结合峰位置。接下来,就需要对峰位置进行注释,获得这些峰结合在了哪些基因上,位于基因的上下游还是内部,以便了解目标蛋白的功能。
那么,怎样对ChIP-seq峰的结合位置进行注释呢?本篇简介一个非常好用的R包,ChIPseeker,能够快速高效地获得预期结果。
1.输入数据准备
输入数据就是记录ChIP-seq峰位置的bed文件,这是ChIP-seq的常见数据格式,测序公司都会给提供的。如下示例,是在人类细胞中进行的ChIP-seq试验,经下机处理并和人参考基因组hg38比对后获得的bed文件。
bed文件中包含5列信息,以tab键分隔。第1列为峰值所处的染色体id,第2-3列为峰值在该染色体中的碱基位置,第4列为峰的id,第5列为信号强度。用Excel打开bed文件就是长这样。
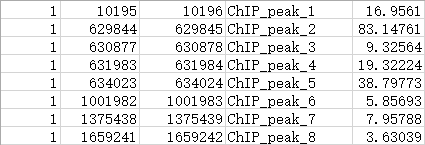 bed文件内容格式
bed文件内容格式
2.ChIPseeker安装
ChIPseeker包可以使用Bioconductor进行安装。
!!!**************************************************
#安装
BiocManager::install("ChIPseeker")
#加载
library(ChIPseeker)
!!!**************************************************
3.准备或构建基因组注释库
为了对ChIP-seq的峰位置进行注释,查看蛋白主要结合到基因组的哪些区域,首先需要准备对应物种的基因组注释库。
例如,上述给定的示例bed文件来源物种是人,因此就需要准备人类参考基因组注释库。有些R包,例如org.Hs.eg.db中提供了已经构建好的人类参考基因组注释库,可以直接调用。此外,也可以通过GenomicFeatures包,读取基因组注释文件gff3本地构建。
推荐通过GenomicFeatures包本地构建注释库,因为绝大多数物种都是没有现成的库可用的,方法灵活。而且,像人类参考基因组也是有很多版本的,可能已知的库和自己使用的基因组版本不匹配,用错了就尴尬了,为了避免万一还是推荐从头构建一下。
!!!**************************************************
#可以使用现有的库,如org.Hs.eg.db包的“org.Hs.eg.db”
library(org.Hs.eg.db)
#或者自己构建一个,指定gff3注释文件即可
library(GenomicFeatures)
spompe <- makeTxDbFromGFF('Homo_sapiens.GRCh38.98.gff3')
!!!**************************************************
4.注释ChIP-seq的峰位置(单个bed文件的注释)
好了,接下来就展示ChIPseeker注释ChIP-seq峰的全过程。
已知上述ChIP-seq的bed文件获取过程中,比对的参考基因组来自hg38,因此首先通过本地准备的人类参考基因组hg38的gff3注释文件,构建本地注释库,随后读取ChIP-seq的bed文件进行注释。
!!!**************************************************
#推荐手动构建TxDb注释文件
spompe <- makeTxDbFromGFF('Homo_sapiens.GRCh38.98.gff3')
#读取chipseq峰的bed文件
peak <- readPeakFile('ChIP-seq.bed')
#注释,TSS的范围可自定义
peakAnno <- annotatePeak(peak, tssRegion = c(-3000, 3000), TxDb = spompe)
#输出结果
write.table(peakAnno, file = 'peak.txt',sep = '\t', quote = FALSE, row.names = FALSE)
!!!**************************************************
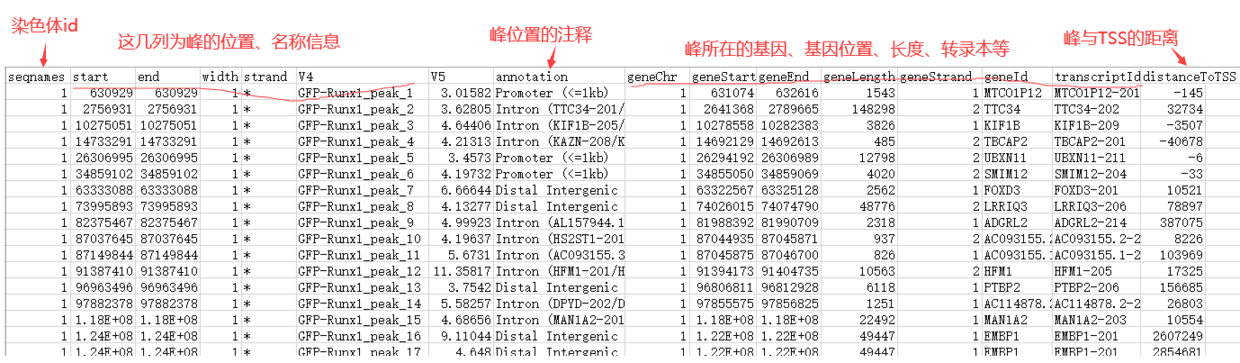 ChIPseeker输出表格内容展示
ChIPseeker输出表格内容展示
结果中,注释了ChIP-seq峰结合的染色体位置、区段、基因名称、基因位置等信息。
对于基因组区域,基因启动子区、外显子或内含子区均有。后续就可以根据这些信息作一些筛选,例如ChIP-seq试验使用的蛋白如果是转录因子,那么通常就是在启动子区发挥作用,只关注启动子区的结合位点即可。
同时,ChIPseeker包中自带了一些可视化函数,可以进行一些简单的统计帮助我们了解数据分布,例如ChIP-seq峰所在基因不同位置的数量占比等。
!!!**************************************************
plotAnnoBar(peakAnno)
vennpie(peakAnno)
plotAnnoPie(peakAnno)
plotDistToTSS(peakAnno)
!!!**************************************************
 一些简单统计图
一些简单统计图
5.注释ChIP-seq的峰位置(多个bed文件的批量处理)
很多情况下,ChIP-seq试验不止作了一次,可能存在多个bed文件有待处理。如果这时,一个个分别注释就会略显繁琐。好在ChIPseeker也提供了批量注释的方法,如果有多个bed文件,可以放在一个list里面统一执行,更加高效。
参考以下示例,两个bed文件的注释,参考基因组仍然是人类hg38,因此继续使用上文构建好的库执行。
!!!**************************************************
#读取chipseq峰的bed文件
peak1 <- readPeakFile('ChIP-seq1.bed')
peak2 <- readPeakFile('ChIP-seq2.bed')
peaks <- list(peak1 = peak1, peak2 = peak2)
#注释
peakAnnoList <- lapply(peaks, annotatePeak, TxDb = spompe, tssRegion = c(-3000, 3000), addFlankGeneInfo = TRUE, flankDistance = 5000)
#分别输出结果
write.table(peakAnnoList[1], file = 'peak1.txt',sep = '\t', quote = FALSE, row.names = FALSE)
write.table(peakAnnoList[2], file = 'peak2.txt',sep = '\t', quote = FALSE, row.names = FALSE)
#可视化,两个可以放在一起比较
plotAnnoBar(peakAnnoList)
vennpie(peakAnnoList[[1]])
plotAnnoPie(peakAnnoList[[1]])
plotDistToTSS(peakAnnoList)
!!!**************************************************
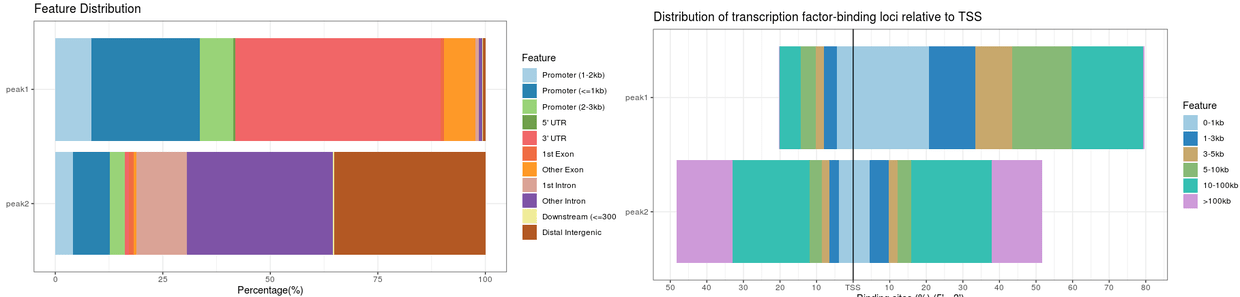 多组比较统计图
多组比较统计图
输出表格略,内容和单个运行的结果是一致的。对于输出结果图,则是将两组放在一起比较的图。
!
!
!
注:!!!*******之间为R脚本内容
搜索微信公众号“纪伟讲测序”- 本文固定链接: https://maimengkong.com/zu/1311.html
- 转载请注明: : 萌小白 2022年12月16日 于 卖萌控的博客 发表
- 百度已收录
