此前我们已讲解许多关于RNA甲基化相关实验设计和研究思路套路,今天,我们来点新干货,从生物信息学分析的角度来独家解析MeRIP-Seq数据的分析利器——exome Peak。通过exomePeak包能够进行RNA甲基化位点peak鉴定、peak定量以及差异分析,一起来看看这款经典软件的内部原理吧。
关于exomePeak
exomePeak是目前主流的MeRIP-seq(m6A-seq)分析工具。最初版本是由孟佳课题组基于MATLAB语言编写的,之后的新版本采用R语言编写,使得这款软件应用更加简便。该工具主要用于组内Peak鉴定、组间差异Peak分析,以揭示RNA甲基组转录后调控的动态。exomePeak支持多个生物学重复、内部自动去除PCR重复和多重比对序列,peak鉴定基于外显子的连接位点(RNA甲基化位点),而非全基因组进行考虑,增强了peak鉴定的准确性,使得该工相比MACs等工具更适合用于m6A-seq数据分析。
exomePeak软件的核心算法
核心peak检测的算法是基于Przyborowski和Wilenski的两个Poisson分布均值比较的方法(C检验)。更具体地说,我们假设落在基因外显子窗口w=[w1,w2]上的测序reads数量服从Poisson分布:
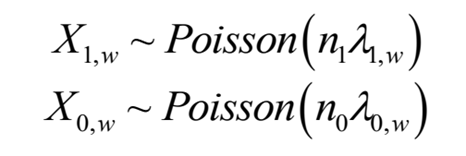
Tips:泊松分布:当一个小的概率固定(λ)的独立重复实验,重复次数很大(n)的时候,成功的概率趋近于固定值nλ,成功的次数满足泊松分布。
在这里,测一条read落在基因组上的哪个区域可以看做一个随机事件(虽然实际上并不随机),正好落在窗口w内的概率很小并且是固定的。当测序reads数很大的时候,落在区间内的概率是固定的。那么落在窗口w上的reads数量就该满足泊松分布。
其中,X1,w和X0,w是IP样本和 Input对照样本比对到窗口w=[w1,w2]内的reads数量,n1和n0是各个样本的reads总数(或测序深度),参数λ1,w和λ0,w分别表示IP和Input样本测序reads落入窗口w的概率。接着,我们来检验一下这些假设:
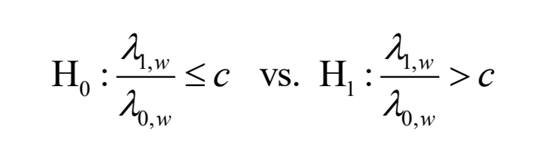
其中c >1是可由用户指定的IP/Input倍数变化阈值。在给定X1,w+X0,w=k的条件下,X1,w的条件分布遵循一个在k次随机试验中成功概率为p的二项分布,概率p定义为:
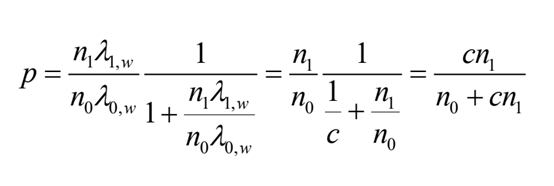
其中c =λ1,w/λ0,w。当p值小于用户定义的显著性水平α时,该条件检验(C-test)拒绝H0假设
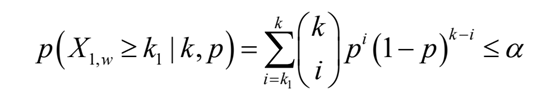
当n1和n0是总reads数时,得到的p值反映了窗口w相对于整个转录组reads的富集倍数(IP vs Input)的显著性。或者,如果n1和n0被设置成是在特定基因外显子区域内的总reads数,则相应的p值将代表窗口w相对于特定基因的富集倍数的显著性。然后用Fisher或Stouffer的方法将两个p值结合起来,以确定转录组范围或基因特异性富集倍数的意义。
1、exomePeak软件考虑的关键问题和相应设计
exomePeak是一个用于检测MeRIP-seq数据中转录后RNA修饰位点的R软件包,使用exomePeak分析可以解决转录组的异质性问题,大大简化了MeRIP-seq数据的分析流程。
输入和输出:exomePeak软件的输入是IP样本 bam文件、对照组Input样本bam文件和基因组注释gtf文件。它以bed12的格式输出外显子区域鉴定的peak。bam文件最好可以通过一些转录组比对软件(如Tophat)进行比对,基因组gtf注释文件应包含用户感兴趣的所有转录本。它可以是从数据库如Illumina iGenome、UCSC Table Browser获得基因注释,或从cufflinks等程序中鉴定出的新转录本。当前版本exomePeak不扫描基因间或内含子区域的peak,这些peak可能属于所提供的gtf文件中未明确定义的转录本。
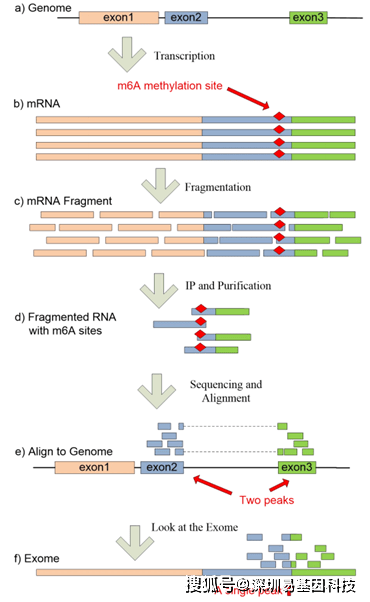
转录组异质性:exomePeak通过直接使用exome避免了异构体水平的差异(图S1)。它可以自动连接由内含子分隔的富集区,并将它们整合为单个 peak。这样的peak在现有的ChIPseq 的peak calling程序中通常会被鉴定为的两个单独的peak。例如,FAM75A1在hg19 上有两个非联合异构:NM_001085452 (chr9: 39355698-39361954)和NM_001085452 (chr9: 39884974-39891205),这两种异构体将分别被单独处理和鉴定。理想情况下,在经过PolyA磁珠富集后,应该只剩下很少的Pre-mRNA,这样就不会鉴定到基因间隔区和内含子区域的peak。对于含有Pre-mRNA的特殊文库,可能需要先使用转录组de novo组装软件,如Cufflinks,在使用exomePeak进行peak鉴定之前先对所有现有的转录本进行识别。
位移大小:由于使用非链特异性文库和单端测序两种统计模式,比对到“-”链的read需要位移。read位移的大小可以由用户设置,一般默认为片段长度的一半(图S2)。
特定位置read数量:我们使用一个大小为w=2f-r的滑动窗口,其中f是片段长度,r是reads长度。窗口大小可以覆盖所有read,其起始点可能与甲基化位点有关。利用span为w的“smooth”函数对特定位置reads密度进行计算。“smooth”函数可以算作一个动态平均函数,下面以span等于5为例,计算各位点reads密度:
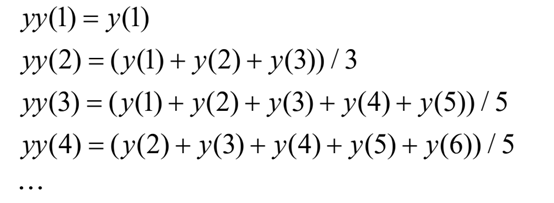
连接read,连接富集区,跳跃外显子的peak: exomePeak软件以与ChIPseq spp软件用相同的方式处理短read。它仅适用于reads的起始位置,并假设read将跨越属于同一基因的相邻外显子。在大多数情况下,属于同一基因的两个相邻的富集区域可以自动连接并报告为一个single peak;然而,当甲基化peak跨越一个跳跃外显子时,由于它们之间的外显子未富集,它将被报告为两个single peak。这本质上是一个转录本可变剪接混淆的问题,而exomePeak没有完全解决这个问题。
2、FDR:错误发现率是通过交换IP和输入样本来计算的,在结果报告中Bed12文件的第9列出现。 FDR是通过比较IP样品和Input样品中的总富集面积(peak的总长度)来计算的。交换IP和Input样本后,exomePeak将在显著性水平FDR<0.05处正确输出0个peak。这与ChIP-seq的许多其他软件工具(如报告q值的MACS2)有着根本的不同。对于 MACS2,如果交换 IP和Input样本,它仍然会在FDR<0.05 时输出超过30k的假阳性peak。
 图S1
图S1
背景估计:exomePeak提供两种模式来估计X0,w
‘p’方法,或称‘positional’方法,它使用Input对照样本中相应窗口区域的实际read计数。由于该方法过于灵敏,可能会导致许多假阳性peak。
“mga”方法,或“最小基因平均值”方法,它使用该基因的平均tag密度,该窗口中的tag密度应该低于该基因的平均密度,否则与“p”相同。当 PCR 扩增偏好或 3' 偏好较少时,这种方法更为保守且效果更好。尽管如此,它在灵敏度方面不及‘positional’方法。
内含子和基因间隔区的peak:exomePeak不扫描内含子区域,也不报告任何内含子peak。根据目前的RNA表观基因组测序方法,在PolyA提纯后,应该只剩下很少的Pre-mRNA,在理想情况下,不应该有任何read比对到内含子或基因间隔区。然而,在实际操作中,m6A甲基组数据中确实存在着内含子peak和基因间隔区的peak,这主要是由于以下两个原因:
首先,在新的转录本上有m6A甲基化位点,相对于不完整的基因注释数据库,它们被认定为是内含子或基因间隔区。特别是对于研究较少的生物体来说,这是非常常见的。一般来说,如果有已知的感兴趣的转录本,内含子的peak是可以忽略的,因为它们与任何已知的转录本或功能无关。但是,对于对未知的转录本感兴趣的用户,我们建议用户先运行转录组组装软件,然后将未知的转录本与现有的转录本相结合,之后再运行exomePeak进行peak calling。这样,内含子peak就可以与新发现的转录本联系起来。注意到exomePeak可以直接获取由 Cufflink和 Cuffcompare生成的gft格式的基因注释文件,这有助于一些潜在分析的进行。
其次,在数据中可能存在来自Pre-mRNA的m6A甲基化位点,也可能存在来自其他生物的污染的m6A甲基化位点。即使在PolyA筛选后,样品中仍可能含有少量的Pre-mRNA。因此,潜在的污染可能导致内含子区域的read的产生,内含子区域约占人类基因组的98%。由于这些RNA通常作用不大,我们选择在exomePeak中忽略它们。对于需要考虑这些RNA的特殊情况,有必要在如上所述的peak检测之前运行转录组组装软件。
 图S2
图S2
3、过滤PCR duplicates产物
一般来说,duplicates的程度高度依赖于tag密度。tag密度越大,预期的duplicates片段就越多。去除MeRIP-Seq中的重复片段read应该对低表达基因有益,但对高表达基因却不友好。由于我们一般更关心假阳性错误而不是假阴性错误(宁愿错过真正的peak也不将希望将假peak当作真的),所有duplicate read都使用 Samtools rmdup函数过滤。这种类型的过滤将避免由于PCR 产物引起的假阳性peak,但可能会错过高表达转录本上的peak。解决这一问题,可能需要更先进的PCR技术。
考虑长度为n/ 2bp的窗口,其中n是可能的唯一reads的最大数量。实际落入该区域的reads总数为m,唯一reads数x遵循以下分布

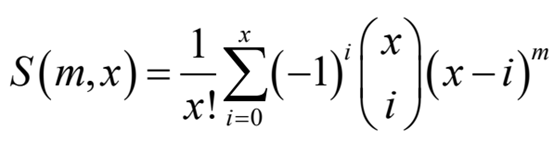
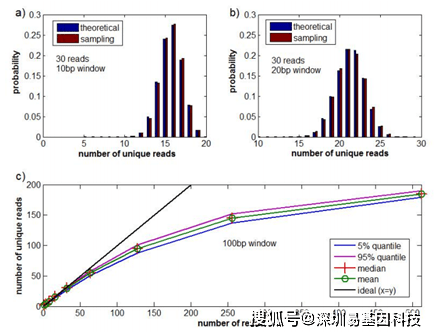
是第二类Stirling数,表示将m个对象的集合划分为x个非空子集的方法数。删除duplicate read后的read计数可以基于此进行更正。特殊read的实际数量可以通过特殊read的平均数量来估计,虽然分析难以处理,但可以通过Monte Carlo抽样方法轻松计算(图S3)。
 图S3
图S3
4、与 ChIPseq Peak Calling 软件的比较
基于外显子方法的exomePeak已针对RNA表观基因组测序数据分析进行了优化,并具有许多独特的设计以适合MeRIP-seq数据处理的功能。表I中提供了exomePeak与MACS2之间的比较。

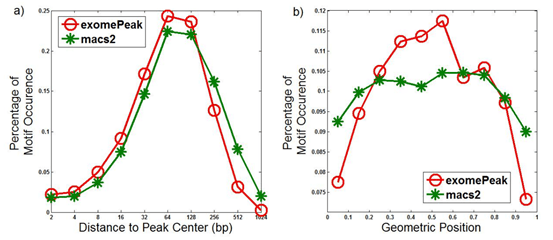 图S4
图S4
有问题可以评论留言交流~
转自:易基因
- 本文固定链接: https://maimengkong.com/zu/1307.html
- 转载请注明: : 萌小白 2022年12月15日 于 卖萌控的博客 发表
- 百度已收录
