此文为Chip-seq报告的解读文件:
以下红字灰色背景为每一小节的结果解读信息
结果解读位于每一小节末尾
1. 工作流程
染色体免疫共沉淀(ChIP)是一种用于研究蛋白质与 DNA 的体内相互作用的经典实验技术。采用特异性抗体将目的蛋白进行免疫沉淀,由此可以把目的蛋白所结合的基因组 DNA 片段也富集下来。通过与高通量测序技术的结合,对 ChIP 后的DNA 产物进行测序分析, 从全基因组范围内寻找目的蛋白的 DNA 结合位点,以高效率的测序手段得到高通量的数据结果。
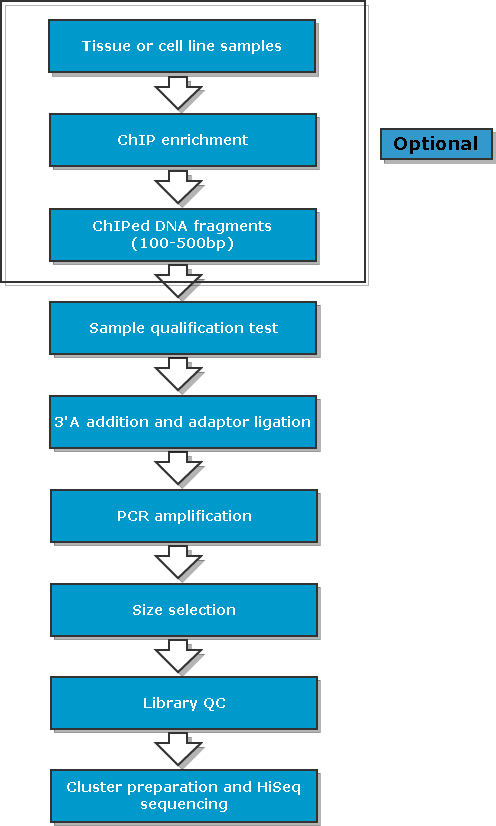
1.1. ChIP 免疫沉淀实验流程
目前主要有两种不同的ChIP 实验方法,大致流程如下(以细胞样品的处理过程为例):
Cross-liking Chromatin Immunoprecipitation (X-ChIP)
-
准备足量的新鲜细胞,每个IP约4x106个细胞,用新鲜的1%的甲醛处理细胞,进行细胞交联。
-
125mM的甘氨酸终止交联,收集细胞。
-
超声或酶解打断染色质,将基因组 DNA 打断至 100-500bp。
-
将抗体(一般为1~5ug)与染色质片段4℃孵育过夜。
-
加入proteinA/G beads进行4℃孵育4-6小时。
-
Proteinase K 解交连。
-
酚氯仿或DNA提取试剂盒提取DNA
-
QPCR 检测或建库测序
1.2. ChIP Sequencing 文库构建流程
-
用qubit 对ChIP片段进行定量检测
-
补齐片段末端,并在3’末端加A尾
-
添加Adapter
-
0.8X AMPure beads去掉多余的Adapter
-
文库PCR扩增
-
1XAMPure beads 去掉多余的primer
-
qPCR测定文库浓度
-
Agilent 2100测定文库片段大小
1.3. 生物信息分析流程
将测序结果与参考基因组比对,比对上唯一位置的序列用于后续标准信息分析及个性化分析。信息分析流程如下:
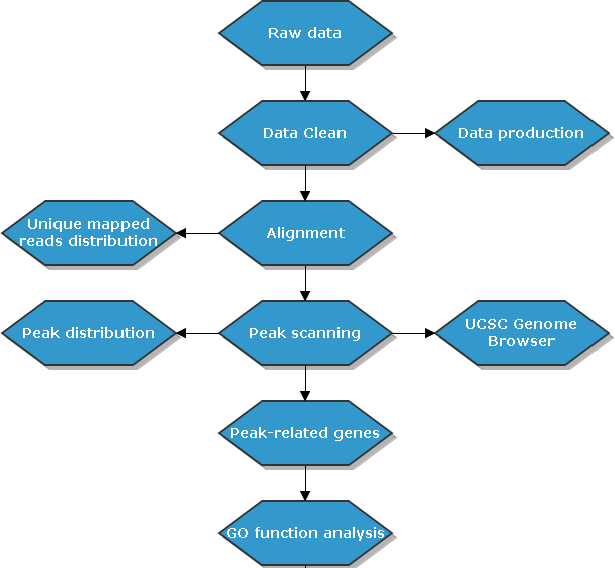
此节内容为Chip-seq基本流程介绍,包括
实验流程
建库流程
分析流程
2. 数据结果及生物信息分析
2.1. ChIP Sequencing 文库质检结果
文库片段质检,ChIP文库的染色质片段在100-500bp之间,建库加入约140bp的接头后,片段应该分布在250-700bp之间为最好。
Fragment Analyzer (FA)毛细管电泳检测:

检测结果汇总:(以下结果中文库大小为 FA 判定结果)
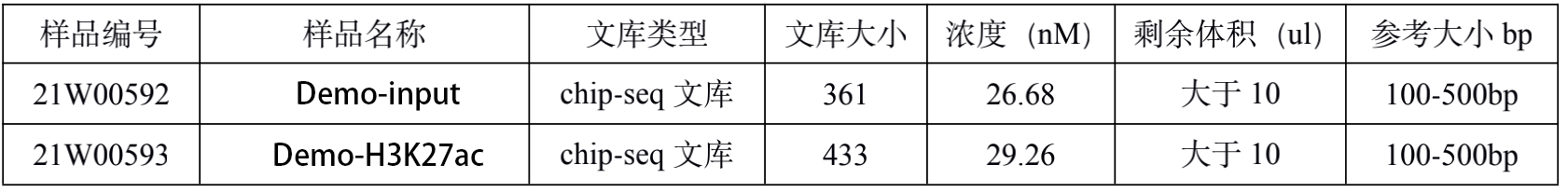
此节内容为Chip-seq文库构建质检结果展示:
图中展示了各个样本文库大小及浓度等信息
2.2. 测序数据质量控制
对原始测序数据及去除接头后的可用数据进行质量评估。
具体的qc报告见:
Results/2.2.QC/qc_Demo-H3K27ac.html
Results/2.2.QC/qc_supplement.html
本节展示了ChIP-seq数据的质量:
比对情况
测序深度
组内重复性
peaks数量、长度分布
peaks中reads的数量百分比
...
2.3. Reads 在全基因组的可视化分布
使用 IGV 软件对 Reads 进行可视化查看,可以查看全基因组任何感兴趣位置的 reads 富集情况,示例如下:
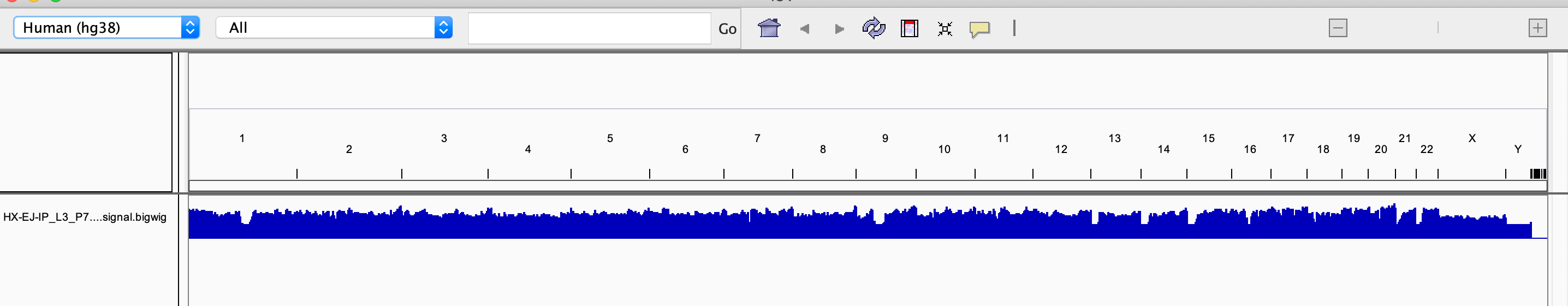
IGV 的安装使用参考: http://software.broadinstitute.org/software/igv/
可视化操作步骤依次是:
在软件的 Genome 选项,基因参考序列 hg38 ;
在软件的 File 选项,上传 要查看染色体 bigwig 文件 以及 narrowPeak文件;
以上文件上传后可查看该染色体任意位置的基因信息及 reads 富集情况。
结果文件 :
表头说明:
Results/2.3.peak_cover/*.narrowPeak表头说明:
| 表头(以下表示第几列) | 说明 |
|---|---|
Column 1
|
seqnames, peak所在染色体 |
Column 2
|
start, peak起始位置 |
Column 3
|
end, peak终止位置 |
Column 4
|
peakname, peak的名字 |
Column 5
|
score, callPeak的置信度分数,结果按照该列进行排名,计算方法为int(-10*log10Pvalue)
|
Column 6
|
strand, 正负链信息 |
Column 7
|
FC, target vs input 的倍数 |
Column 8
|
score, pvalue,计算方法为-10*log10Pvalue
|
Column 9
|
score, qvalue,计算方法为-10*log10qvalue
|
Column 10
|
两个峰最高点之间的距离,示例如图 |
Results/2.3.peak_cover/*.narrowPeak表头说明图示:
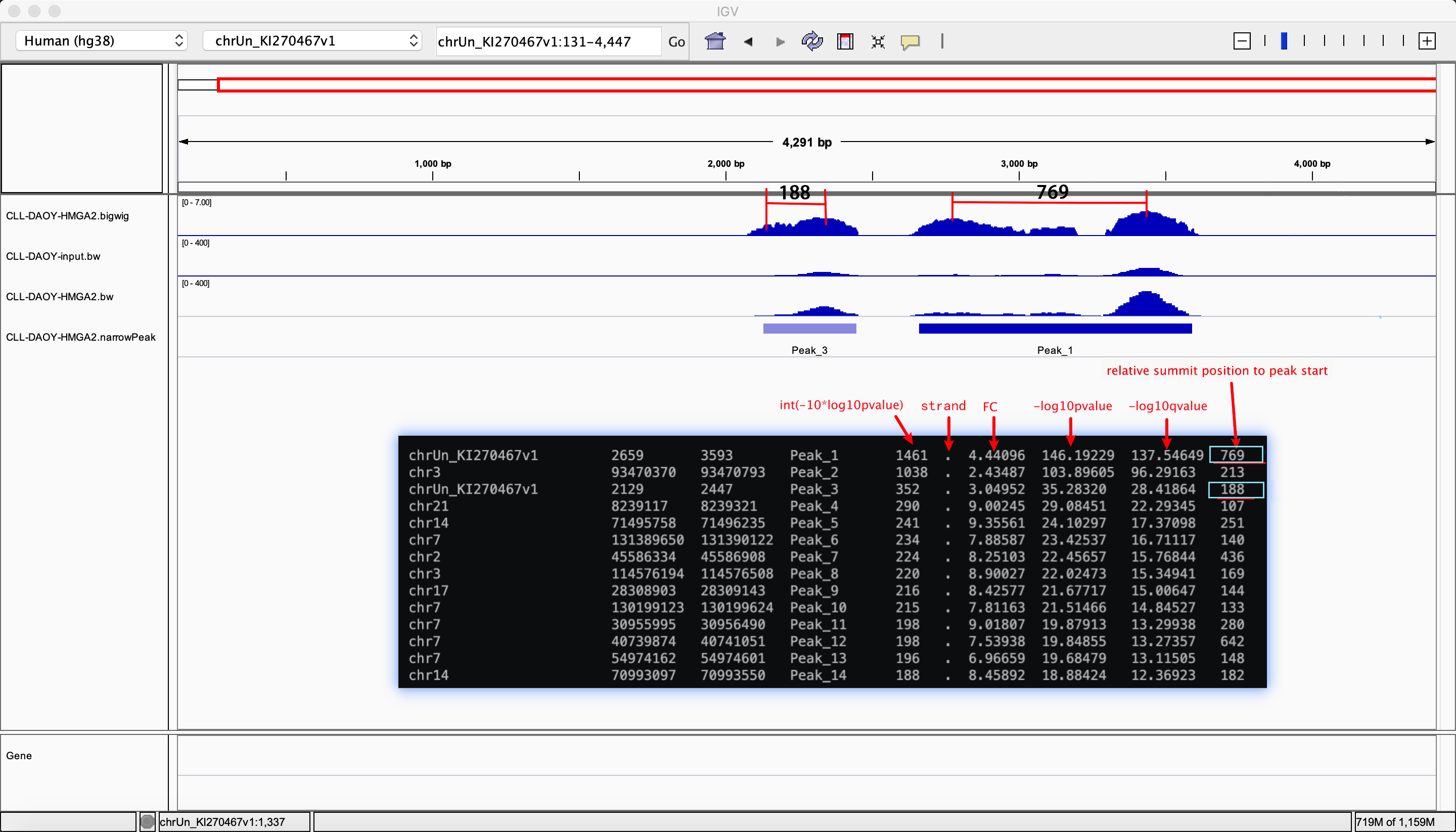
此节内容,对结果进行说明并给出了在IGV中可视化的两个最重要的基本文件:
.bigwig: 测序reads在基因组上的可视化分布结果文件
.narrowPeak: callPeak结果文件
2.4. 全基因组 Reads 富集峰 Peak 鉴定
采用常用 reads 富集峰鉴定软件 MACS 在全基因范围进行 peak 扫描,得到 Peak 在基因组上的位置信息、peak 富集信息等。

图1 全基因组 Reads 富集峰
结果文件:
Results/Demo-H3K27ac.PeakAnno.xls
Results/2.4.peak_scan/Demo-H3K27ac.covplot.pdf
表头说明:
Results/*.PeakAnno.xls表头说明:
| 表头 | 说明 |
|---|---|
seqnames
|
peak所在染色体 |
start
|
peak起始位置 |
end
|
peak终止位置 |
width
|
peak长度 |
strand
|
正负链信息 |
V4
|
同Peak文件第4列,peakname,peak的名字 |
V5
|
同Peak文件第5列,callPeak的置信度分数,计算方法为int(-10*log10Pvalue)
|
V6
|
同Peak文件第6列,与上述strand列一致,表示正负链信息 |
annotation
|
peak注释信息(对于注释到基因上等注释信息的描述) |
geneChr
|
注释基因的染色体信息 |
geneStart
|
注释基因的起始位置 |
geneEnd
|
注释基因的终止位置 |
geneLength
|
注释基因的长度 |
geneStrand
|
注释基因的正负链 |
geneId
|
注释基因的EntrezID |
transcriptId
|
注释基因的转录本名字 |
distanceToTSS
|
被注释Peak距离TSS的距离 |
ENSEMBL
|
注释基因的ENSEMBL名 |
SYMBOL
|
注释基因的SYMBOL名 |
GENENAME
|
注释基因的基本描述信息 |
此节内容包括:
所有Peak的临近基因注释结果文件
callPeak结果在全基因组上的分布情况(高度代表置信度)
2.5. Reads 在 TSS 近端富集强度分析
TSS 转录起始位点近端(0-3kb)与特定的基因转录调控功能有关,统计 reads 在TSS 近端的分布情况。
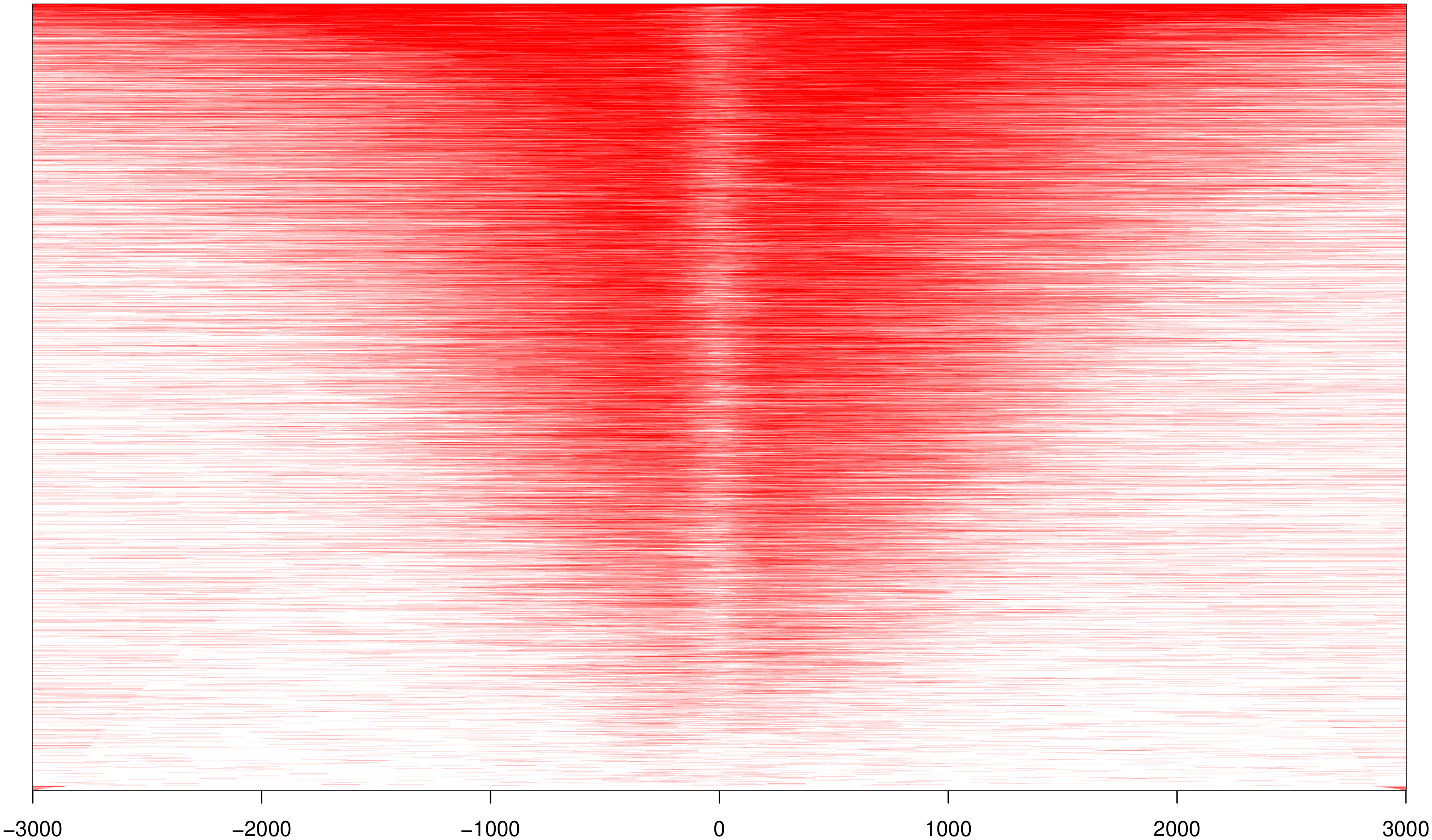
图 2 reads 在 TSS 近端富集强度的分布(热图分布)
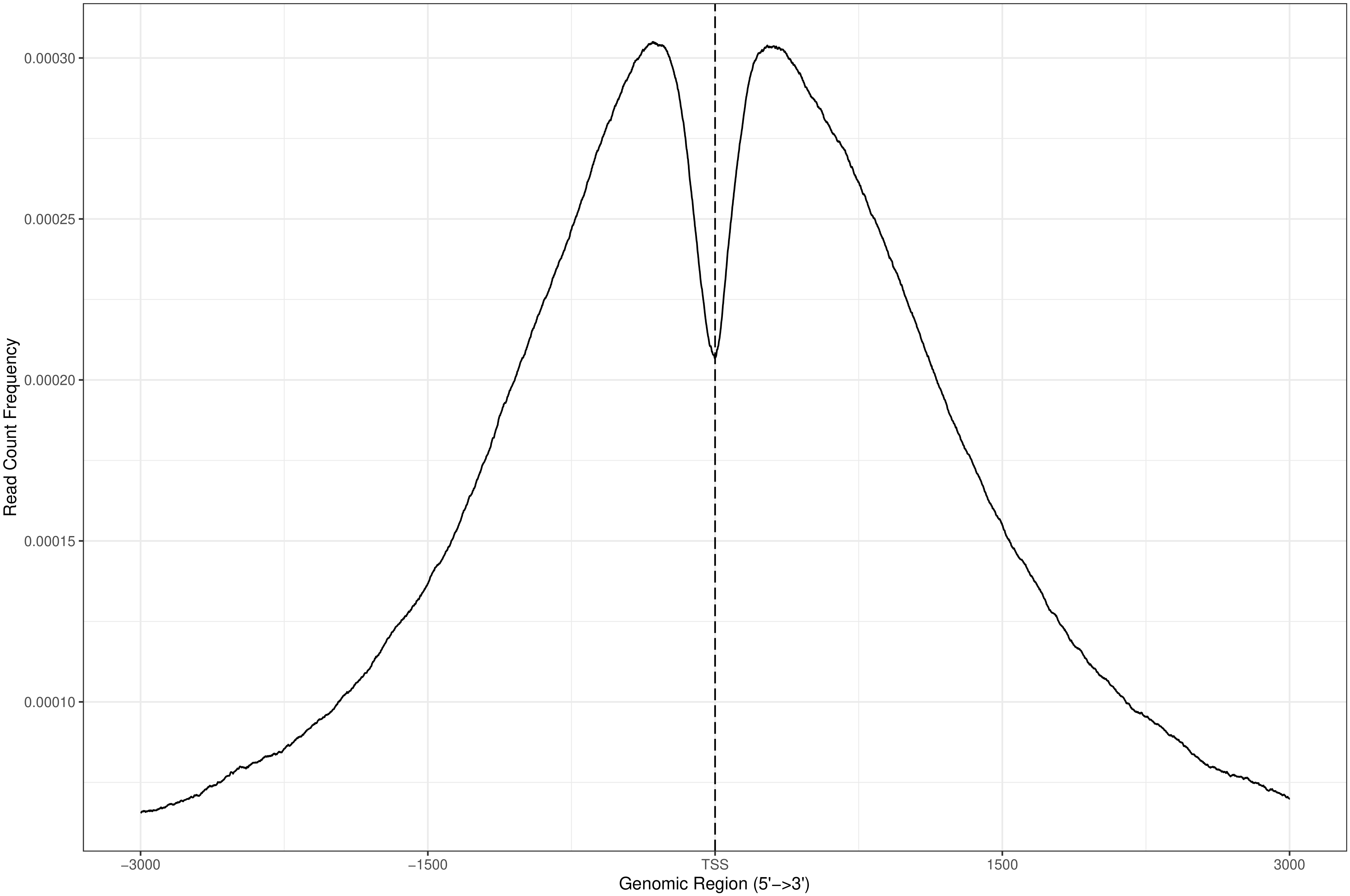
图 3 reads 在 TSS 近端富集强度的分布(峰图分布)
结果文件:
Results/2.5.tss_near/Demo-H3K27ac.tagheatmap.pdf
Results/2.5.tss_near/Demo-H3K27ac.plotavgprof.pdf
此节内容包括:
以TSS为中心向正负拓展3k距离的的Peak富集情况
2.6. Reads 在 TSS 近端及远端富集强度分析
TSS 转录起始位点近端(0-3kb)及远端(10kb以上)的 reads 分布与特定的基因转录调控功能有关,统计 reads 在TSS 近端及远端的分布情况。
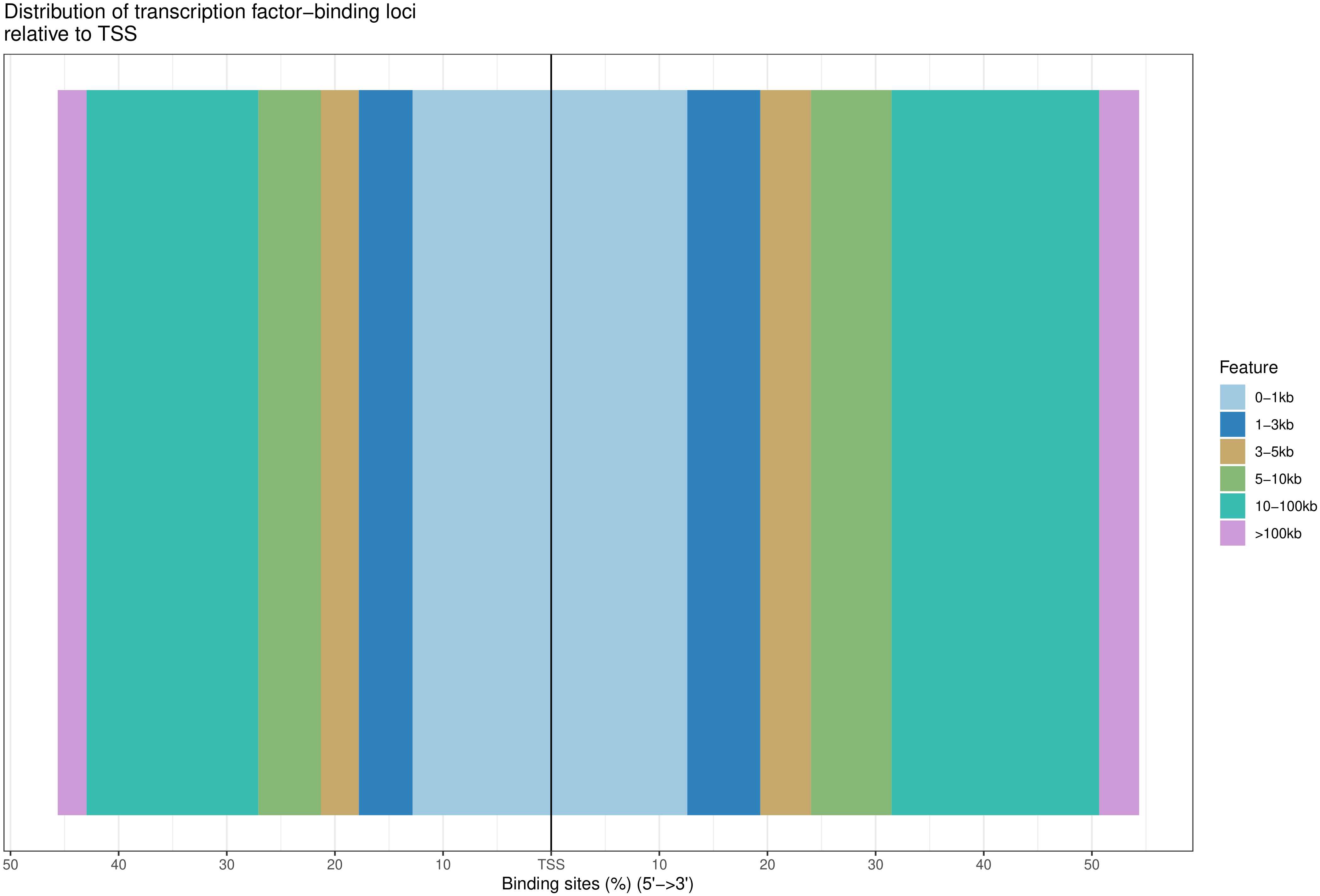
图4 reads 在 TSS 近端及远端富集强度的分布
结果文件:
此节内容包括:
以TSS为中心向正负拓展3k-10k以上距离的的Peak富集情况
2.7. Peak 在基因组上的分布
将 Peak 根据位置信息进行基因组注释基因结构元件,分别统计 Peak 在结构元件(intergenic region、upstream 5K、5`UTR、exon、intron,3’UTR、downstream5k)的数目,并根据其在各个元件上的富集程度,绘制分布特征。
Peak 在基因结构元件上的分布特征:
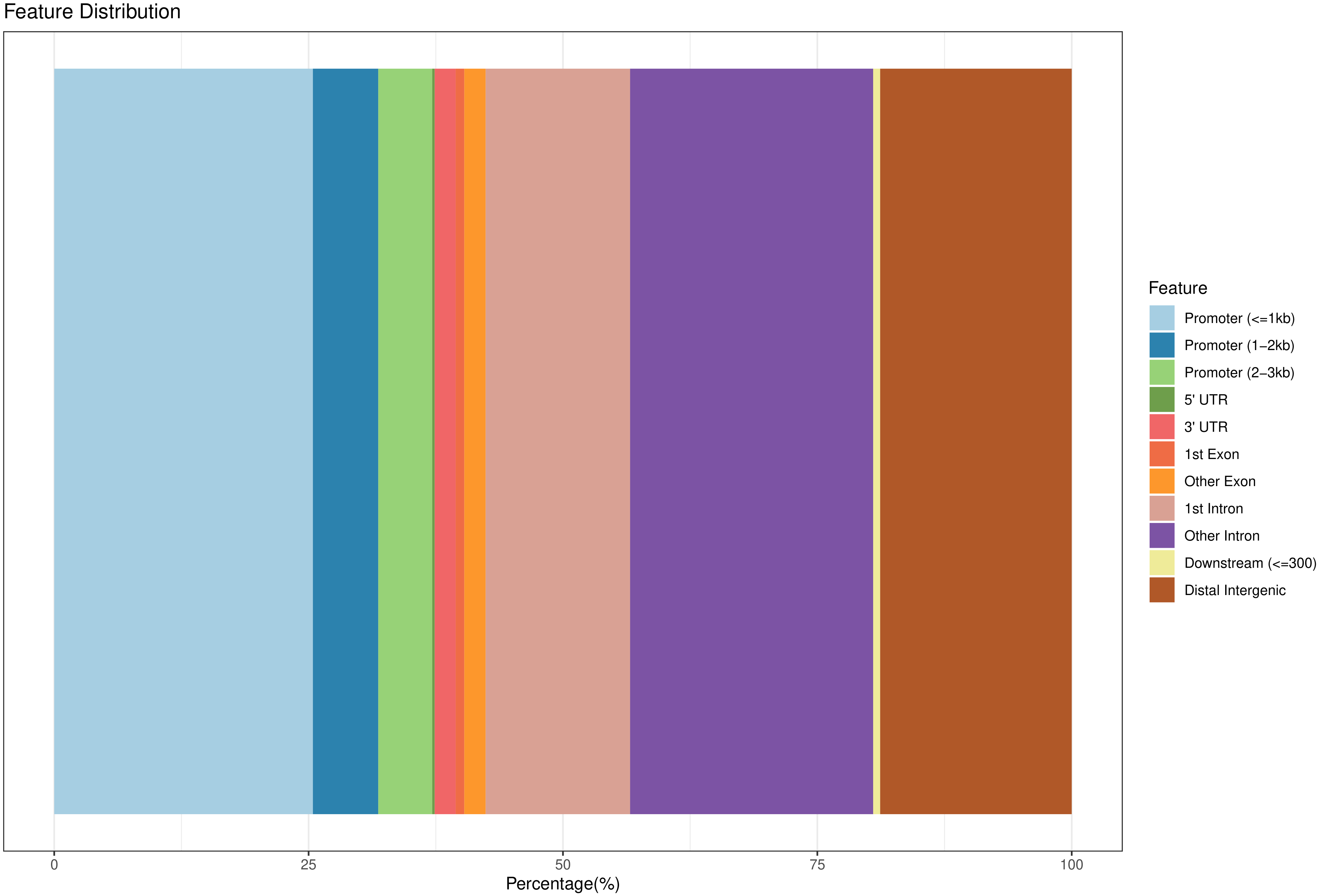
图5 Peak 在基因结构元件上的分布
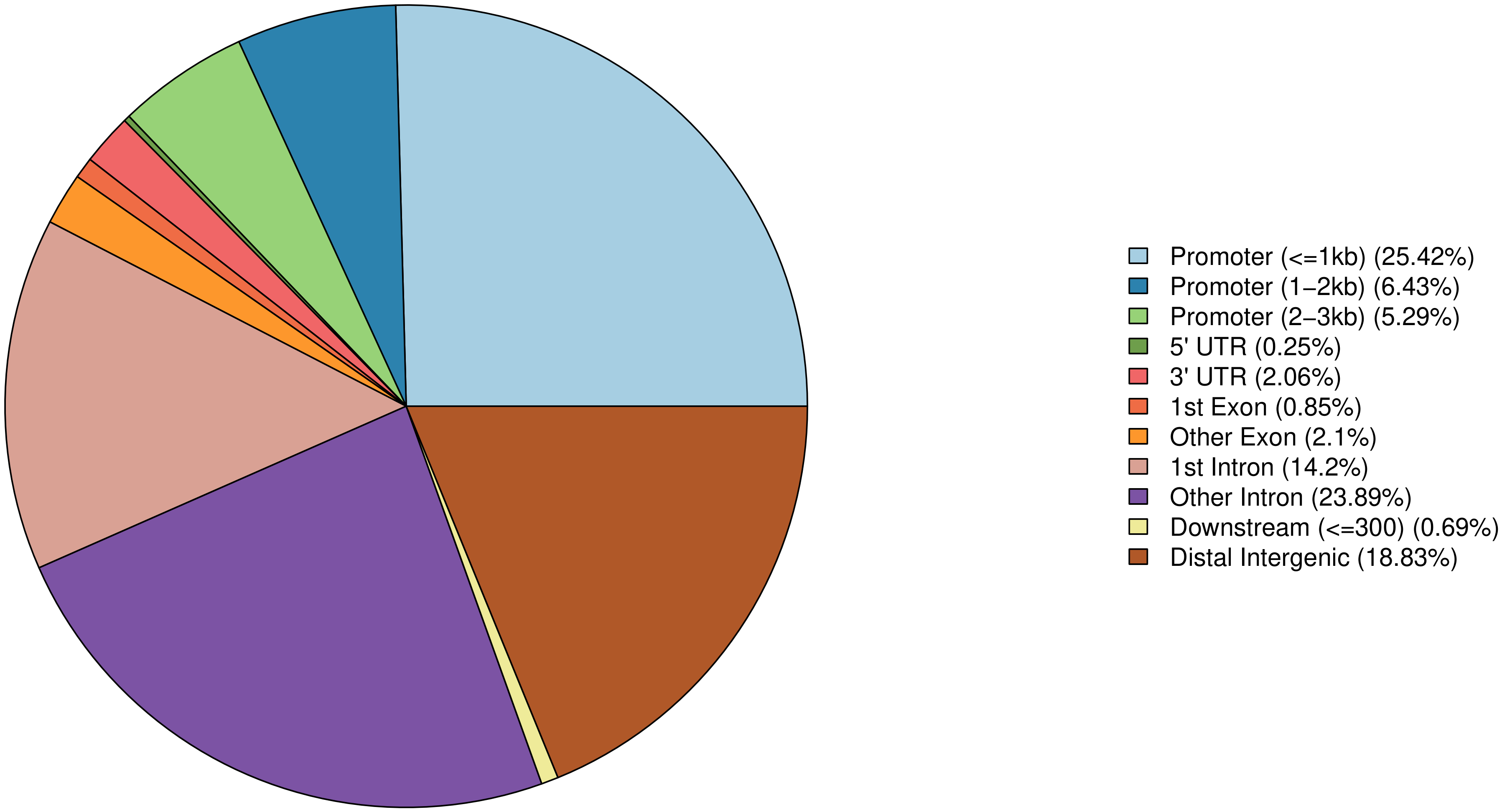
图6 Peak 在基因结构元件上的分布比例
Peak 在各基因结构元件上的交叉分布特征:
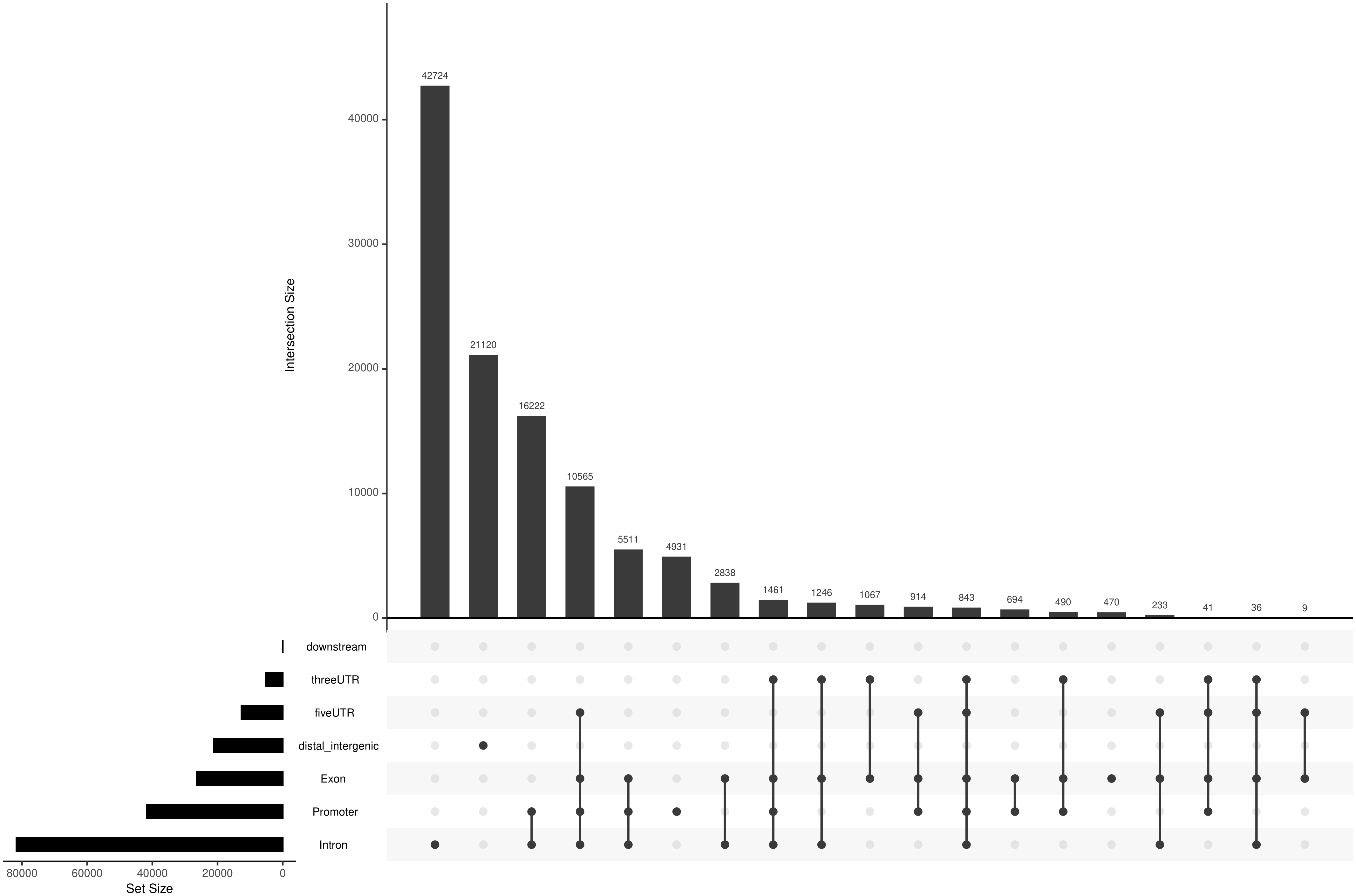
图7 Peak 在基因结构元件上的交叉分布(upsetplot)
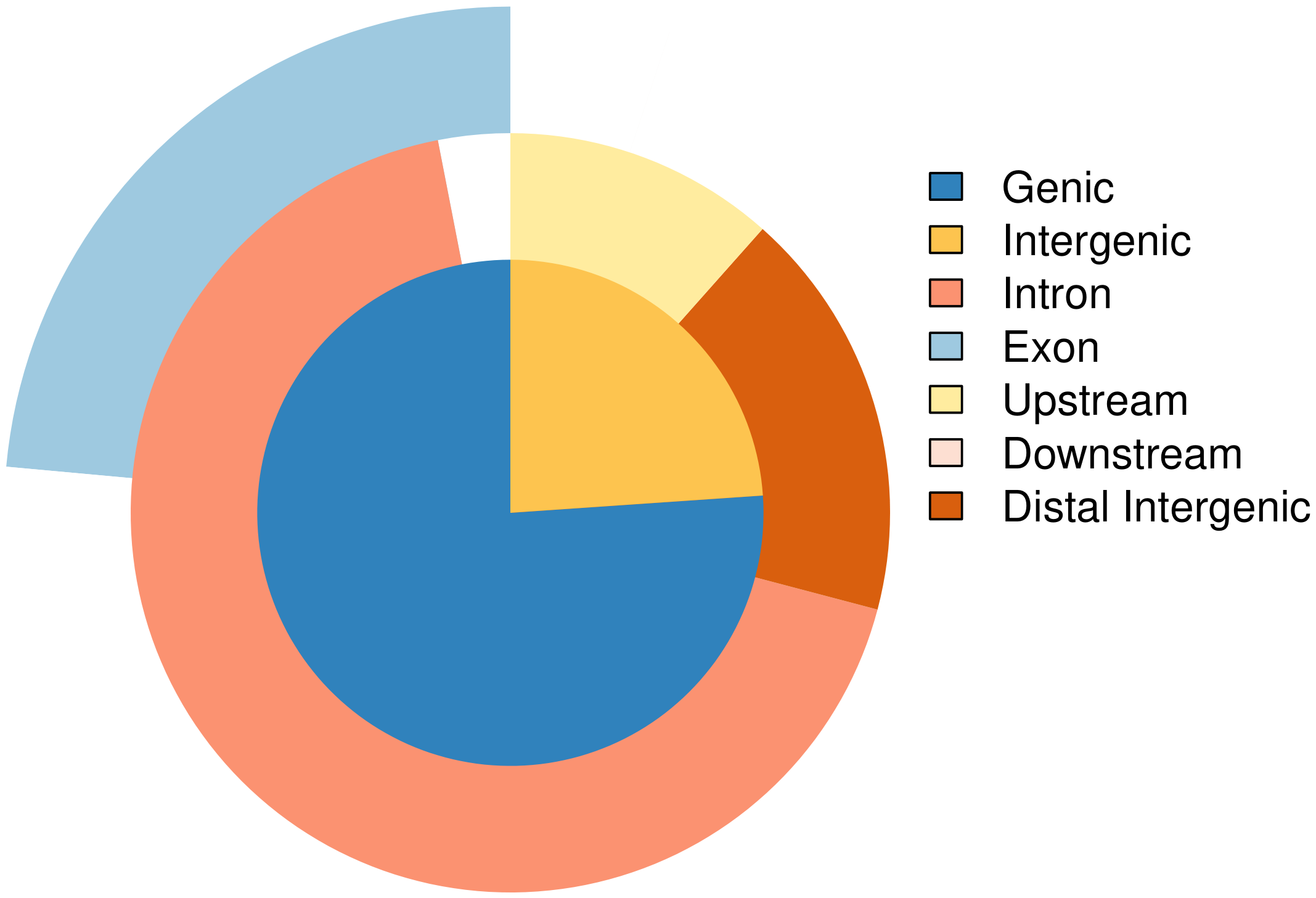
图8 Peak 在基因结构元件上的交叉分布(vennpie)
结果文件:
Results/2.7.peak_dis/Demo-H3K27ac.peakAnnobar.pdf
Results/2.7.peak_dis/Demo-H3K27ac.peakAnnopie.pdf
Results/2.7.peak_dis/Demo-H3K27ac.peakAnnoupset.pdf
Results/2.7.peak_dis/Demo-H3K27ac.peakAnnovinnpie.pdf
此节内容包括:
所有Peak在基因结构元件上的分布特征(即,各个Peak注释到了基因的什么结构元件的比例统计)
所有Peak在各基因结构元件上的交叉分布特征(即,各个Peak注释到的同一个基因,同时分布在多个基因元件的数量统计)
2.8. Peak注释基因的富集分析
我们将前面分析得到的Peak注释基因,进行后续富集分析。
我们根据基因表达量分析得到差异基因之后,必须进一步落到基因的功能上来。对于差异分析而言,往往涉及到成千上万个基因,这会使分析变得很复杂。解决思路是将一个基因列表分成多个部分,从而减少分析的复杂度。为了解决怎么分成不同类,通常会对基因功能进行富集分析, 期望发现在生物学过程中起关键作用的生物通路, 从而揭示和理解生物学过程的基本分子机制。功能富集分析可以将成百上千个基因、蛋白或者其他分子分到不同的通路中,以减少分析的复杂度。另外,在两种不同实验条件下,激活的通路显然比简单的基因或蛋白列表更有说服力。基因功能富集分析首先要构建基因集( gene set,如 GO 和 KEGG 数据库等),也就是基因组注释信息进行分类。然后再把我们的目标基因集(差异基因集或者其他基因集)映射到背景基因集上,注意区分注释与富集。
我们采用 clusterProfiler 软件对差异基因集进行 GO 功能富集分析, KEGG 通路富集分析等。富集分析基于超几何分布原理,其中差异基因集为差异显著分析所得差异基因并注释到 GO 或 KEGG 数据库的基因集,背景基因集为所有进行差异显著分析的基因并注释到 GO 或 KEGG 数据库的基因集。富集分析结果是对每个差异比较组合的所有差异基因集、上调差异基因集、下调差异基因集进行富集。本报告中展示的表格是选取某一个比较组合的富集分析结果,图片是部分富集分析结果。
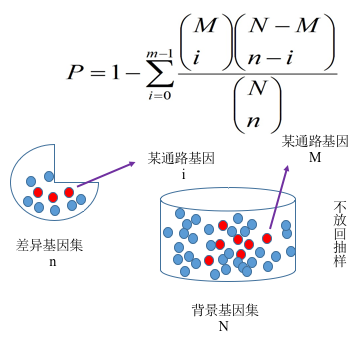
图 9 基因富集分析原理图
2.8.1. 富集分析结果文件
| 结果路径 | 结果说明 |
|---|---|
| GO富集分析结果 |
|
Results/*enrich_*/gene.ego_all-p.adjust1.00.csv
|
GO富集结果列表(所有结果) |
Results/*enrich_*/gene.ego_all-p.adjust0.05.csv
|
GO富集结果列表(按p.adj<0.05筛选后) |
Results/*enrich_*/gene.ego_ALL.csv
|
GO富集结果列表(MF、BP、CC所有结果) |
Results/*enrich_*/gene.GO-*-barplot.p*
|
GO富集分析柱状图 |
Results/*enrich_*/gene.GO-*-dotplot.p*
|
GO富集分析散点图 |
Results/*enrich_*/gene.GO-*-DAG.p*
|
GO富集分析DAG图 |
| KEGG富集分析结果 |
|
Results/*enrich_*/gene.KEGG.csv
|
KEGG富集结果列表(所有) |
Results/*enrich_*/gene.KEGG_significant.csv
|
KEGG富集结果列表(按p.adj<0.05筛选后) |
Results/*enrich_*/gene.KEGG-*-barplot.p*
|
KEGG富集分析柱状图 |
Results/*enrich_*/gene.KEGG-*-dotplot.p*
|
KEGG富集分析散点图 |
| ReactomePA富集分析结果 |
|
Results/*enrich_*/gene.ReactomePA.csv
|
ReactomePA富集结果列表(所有) |
Results/*enrich_*/gene.ReactomePA_significant.csv
|
ReactomePA富集结果列表(按p.adj<0.05筛选后) |
Results/*enrich_*/gene.ReactomePA-*-barplot.p*
|
ReactomePA富集分析柱状图 |
Results/*enrich_*/gene.ReactomePA-*-dotplot.p*
|
ReactomePA富集分析散点图 |
结果文件夹:
-
Pathway1 分析结果文件夹:Results/2.8.enrich_pathway1/
-
Pathway2 分析结果文件夹:Results/2.9.enrich_pathway2/
-
Pathway1 网页预览图:Results/2.8.enrich_pathway1/*-pdf.html
-
Pathway2 网页预览图:Results/2.9.enrich_pathway2/*-pdf.html
说明:
-
Pathway1中对peaks进行基因注释,仅采用临近基因注释。
-
Pathway2中对peaks进行基因注释,需要考虑多个因素,包括注释基因的外显子/内含子,promoter区,也包括peaks两侧可能包含顺式调控元件的区域。
表头说明: (Results/*enrich_*/gene.ego_*.csv GO富集结果列表)
| 表头 | 说明 |
|---|---|
| ID | 对应GO数据库中的ID |
| ONTOLOGY | 分子功能(Molecular Function),生物过程(biological process)和细胞组成(cellular component) |
| Description | GO的描述 |
| GeneRatio | 对应GO 差异基因数 / 能够对应到GO数据库中同类型的差异基因数 |
| BgRatio | 对应GO包含对应物种的基因数 / GO数据库中包含对应物种的基因数 |
| pvalue | 富集分析得到的p-value |
| p.adjust | 校正后的p-value |
| qvalue | 富集分析得到的qvalue |
| Count | 富集基因数目 |
| ENTREZID | 富集基因列表(ENTREZID) |
| SYMBOL | 富集基因列表(SYMBOL) |
表头说明: (Results/*enrich_*/gene.KEGG*.csv KEGG富集、Results/*enrich_*/gene.ReactomePA*.csv ReactomePA富集 结果列表)
| 表头 | 说明 |
|---|---|
| ID | 对应PATHWAY数据库中的ID |
| Description | PATHWAY的描述 |
| GeneRatio | 对应PATHWAY 差异基因数 / 能够对应到PATHWAY数据库中的差异基因数 |
| BgRatio | 对应PATHWAY包含对应物种的基因数 / PATHWAY数据库中包含对应物种的基因数 |
| pvalue | 富集分析得到的p-value |
| p.adjust | 校正后的p-value |
| qvalue | 富集分析得到的qvalue |
| Count | 富集基因数目 |
| ENTREZID | 富集基因列表(ENTREZID) |
| SYMBOL | 富集基因列表(SYMBOL) |
2.8.2. GO功能富集分析
GO(Gene Ontology) 是描述基因功能的综合性数据库,可分为生物过程( biological process )和细胞组成( cellular component )分子功能( Molecular Function )三个部分。 GO 功能富集以 padj 小于 0.05 作为为显著性富集的阈值,富集结果见结果文件。
从 GO 富集分析结果中,选取最显著的 20 个 Term 绘制柱状图进行展示,若不足 20 个,则绘制所有 Term ,按生物过程、细胞组分和分子功能三大类别及差异基因上下调分类画的柱状图。
有向无环图 (Directed Acyclic Graph,DAG) 为差异基因 GO 富集分析结果的图形化展示方式。图中,分支代表包含关系,从上至下所定义的功能范围越来越小,选取每个差异比较组合的 GO 富集结果最显著性前 5 位的 GO Term 作为有向无环图的主节点,并通过包含关系,将相关联的 GO Term 一起展示,颜色的深浅代表富集程度。我们的项目中分别绘制生物过程、分子功能和细胞组分的 DAG 图。
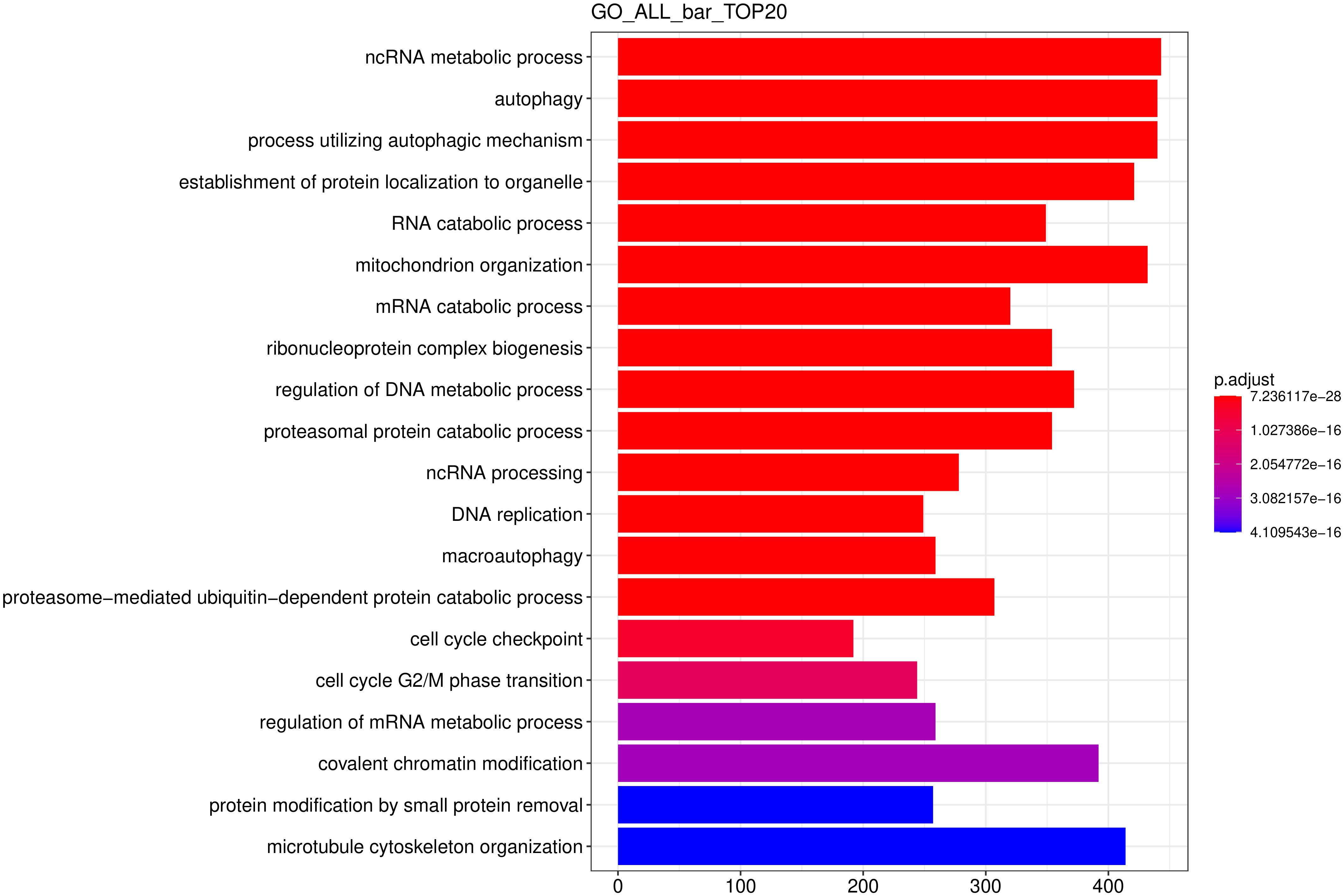
图 10 GO富集分析柱状图
图中纵坐标为GO Term,横坐标为GO Term富集的显著性水平,数值越高越显著
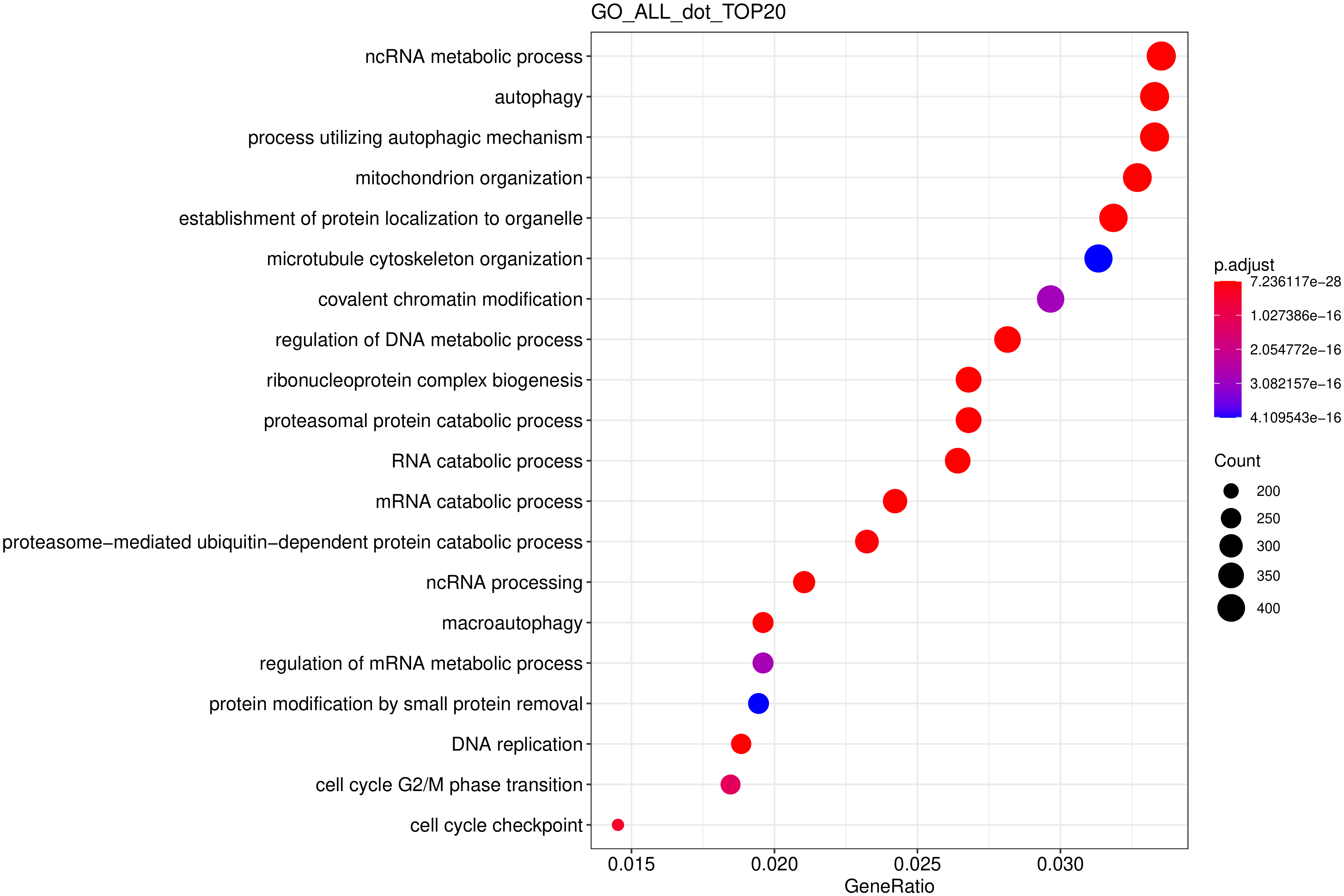
图 11 GO富集分析散点图
图中横坐标为注释到GO Term上的差异基因数与差异基因总数的比值,纵坐标为GO Term
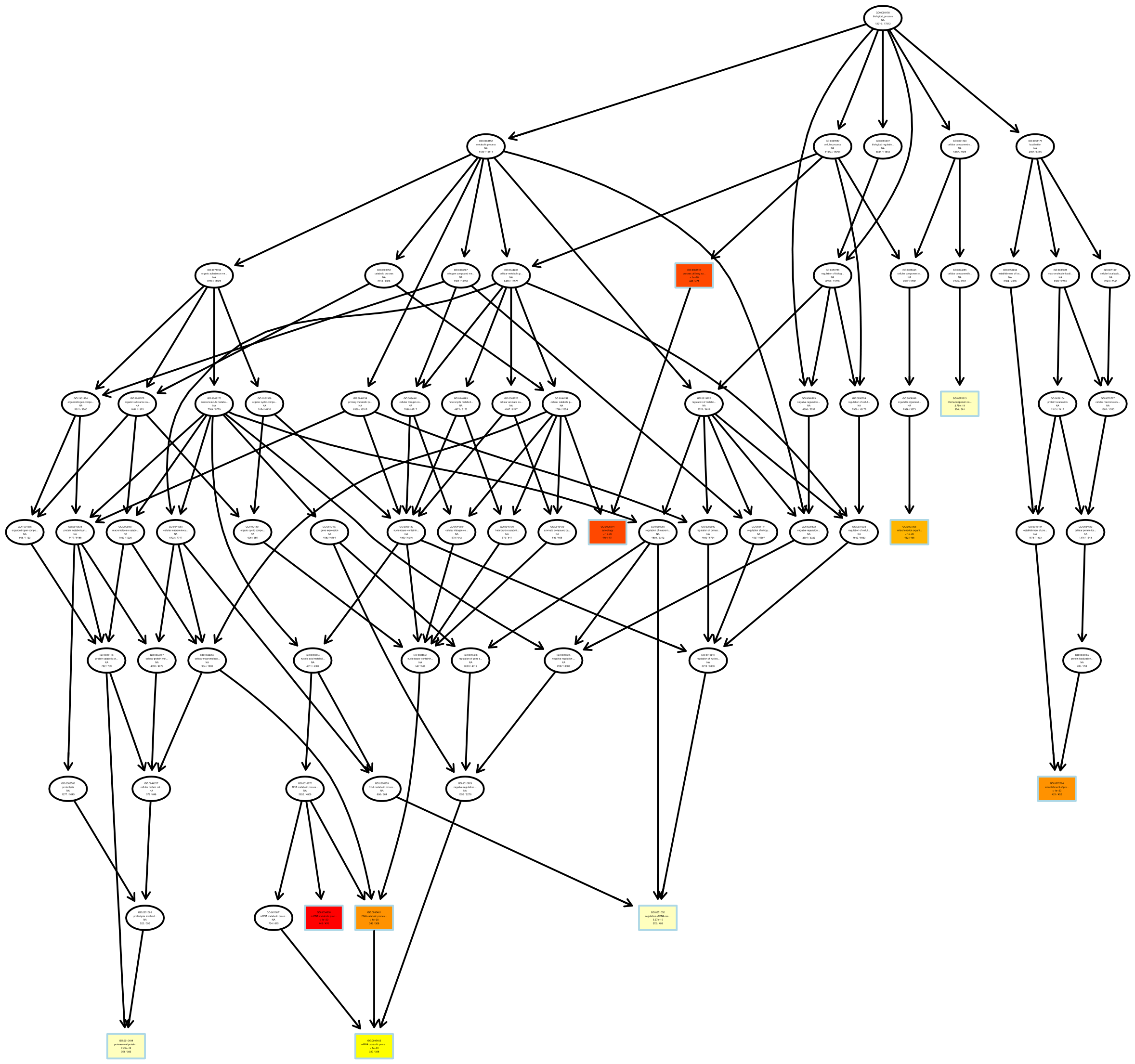
图 12 GO富集分析DAG图
每个节点代表一个GO术语,方框代表的是富集程度为TOP5的GO,颜色的深浅代表富集程度,颜色越深就表示富集程度越高,每个节点上展示了该TERM的名称及富集分析的padj
2.8.3. KEGG通路富集分析
KEGG(Kyoto Encyclopedia of Genes and Genomes) 是整合了基因组、化学和系统功能信息的综合性数据库。 KEGG 通路富集以 padj 小于 0.05 作为显著性富集的阈值,富集结果见结果文件。
从 KEGG 富集结果中,选取最显著的 20 个 KEGG 通路绘制柱状图进行展示,若不足 20 个,则绘制所有通路,如下图所示。图中横坐标为通路富集的显著性水平,数值越高越显著,纵坐标为 KEGG 通路。
从 KEGG 富集结果中,选取最显著的 20个KEGG 通路绘制散点图进行展示,若不足 20 个,则绘制所有通路,如下图所示。图中横坐标为注释到 KEGG 通路上的差异基因数与差异基因总数的比值,纵坐标为 KEGG 通路,点的大小代表注释到 KEGG 通路上的基因数,颜色从红到紫代表富集的显著性大小。

图 13 KEGG富集分析柱状图
图中横坐标为通路富集的显著性水平,数值越高越显著,纵坐标为KEGG通路。
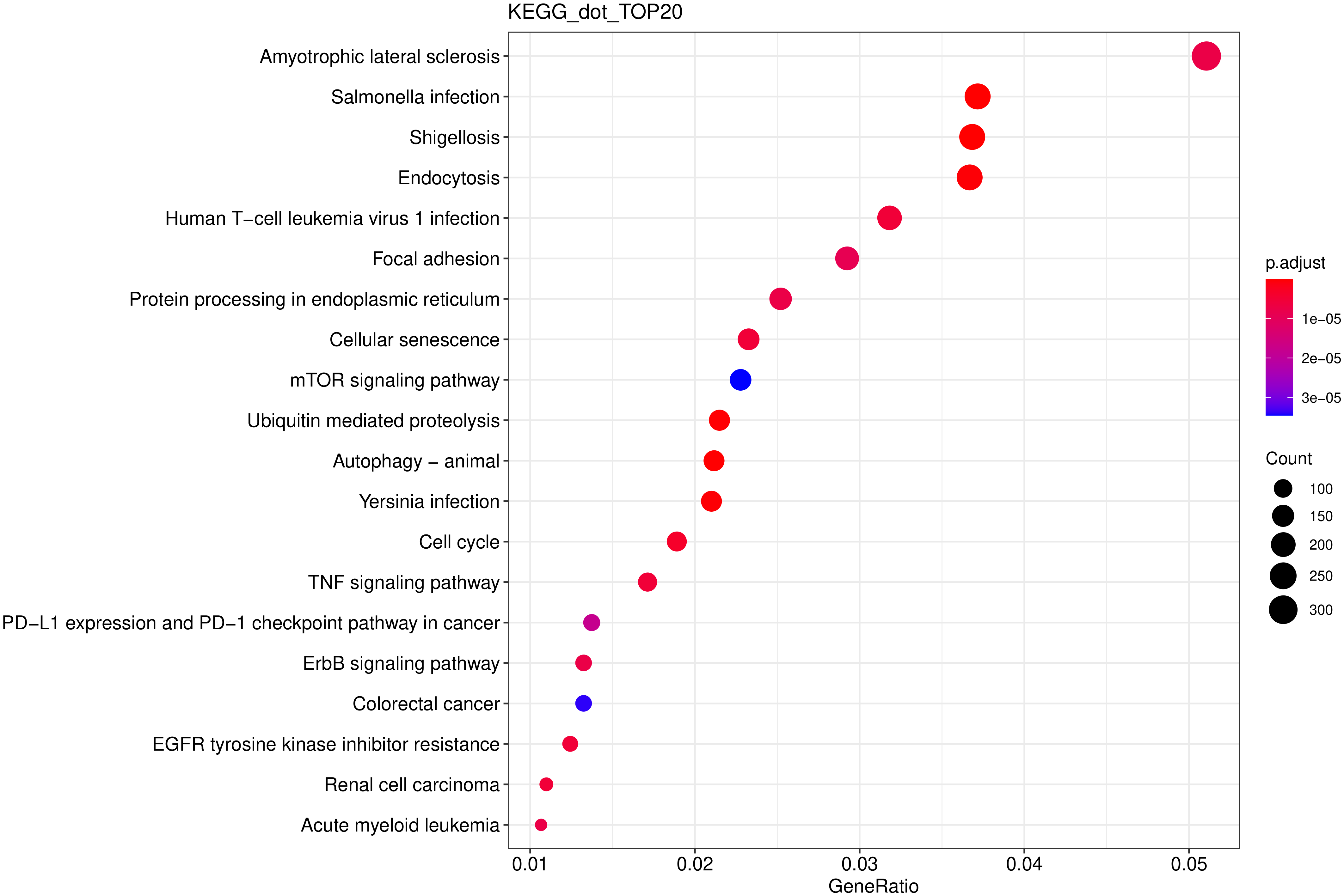
图 14 KEGG富集散点图
图中横坐标为注释到KEGG通路上的差异基因数与差异基因总数的比值,纵坐标为KEGG通路
2.8.4. ReactomePA富集分析
Reactome数据库汇集了人类等模式物种各项反应及生物学通路。Reactome通路富集以padj小于0.05作为显著性富集的阈值,富集结果见结果文件。
以下柱状图与散点图与上一节类似,选取最显著的 20个 富集进行展示,若不足 20 个,则绘制所有通路,如下图所示。
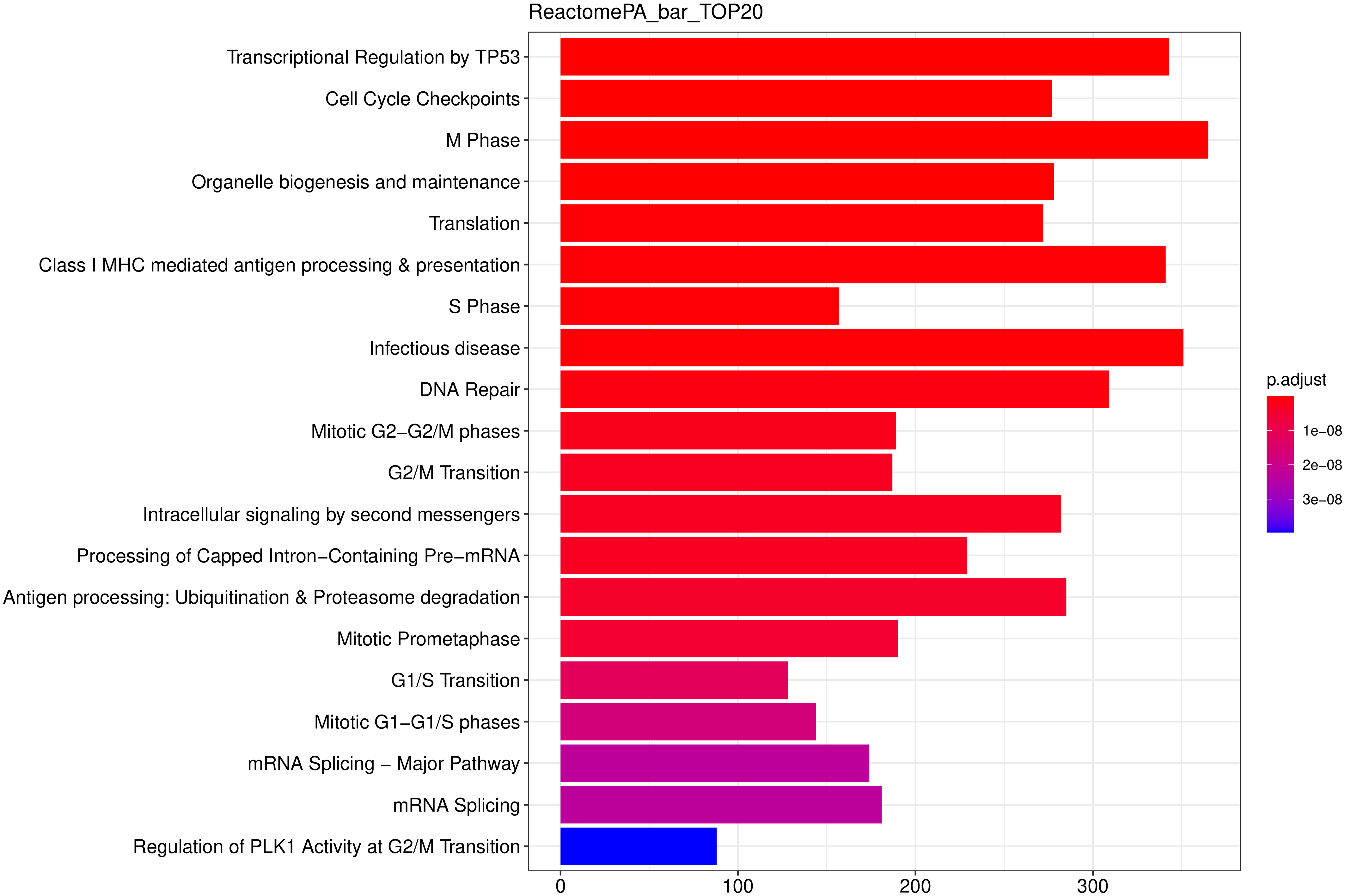
图 15 ReactomePA富集分析柱状图
图中横坐标为通路富集的显著性水平,数值越高越显著,纵坐标为ReactomePA通路。
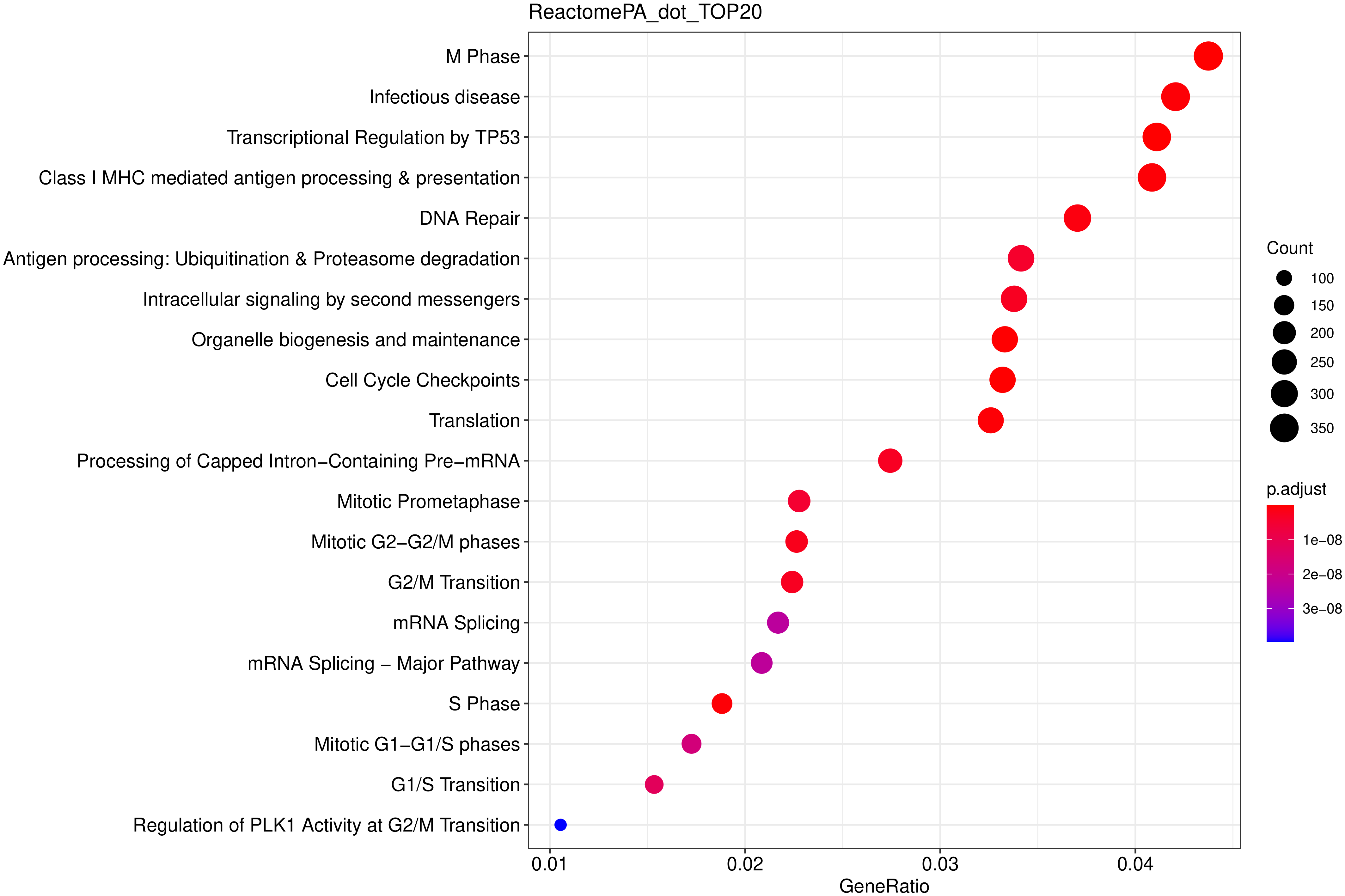
图 16 ReactomePA富集散点图
图中横坐标为注释到ReactomePA通路上的差异基因数与差异基因总数的比值,纵坐标为ReactomePA通路
此节内容包括:
对注释到的基因集的GO富集分析
对注释到的基因集的KEGG富集分析
对注释到的基因集的ReactomePA富集分析
2.9. Peak 区域 Motif 分析
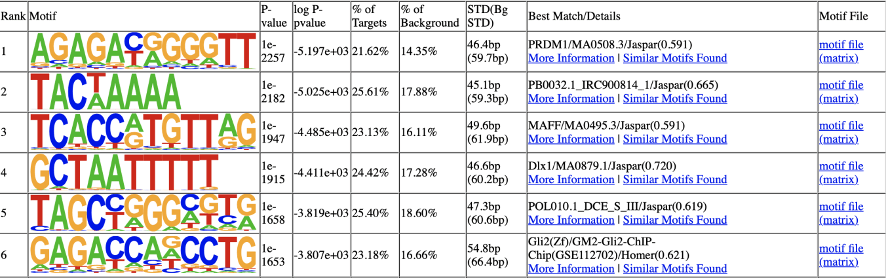
用 Homer 软件对 Peak 区域鉴定 motif 序列;并将得到的 motif 序列与 JASPAR 数据库(JASPAR CORE 2016 database)进行比对,鉴定已知的 motif。
Homer 结果示例:
结果文件:
此节内容为所有Peak区域鉴定 motif 结果,包括:
-
基于JASPAR数据库已知查找结果
-
基于denovo预测结果
- 本文固定链接: https://maimengkong.com/zu/1287.html
- 转载请注明: : 萌小白 2022年11月20日 于 卖萌控的博客 发表
- 百度已收录
