一、 介绍
细菌基因组研究,是通过基因组测序和组装,获得细菌全基因组序列,并对基因组开展结构预测,功能注释、比较基因组学及泛基因组研究。依据研究精细程度不同,分为框架图(也称为草图)、完成图(0gap),下面我们主要介绍框架图的分析内容。
二、 分析流程
基本流程为,建库测序,序列优化,基因组组装,基因及结构预测,功能注释,画图展示。

三、 分析步骤与结果展示
1、测序序列的质控和拼接
2、组装结果评估:把 reads 比对到组装好的基因组序列上, 通过统计组装序列的 GC 含量和 reads 覆盖深度, 总结基因组的 GC偏向性和重复序列情况及污染情况。
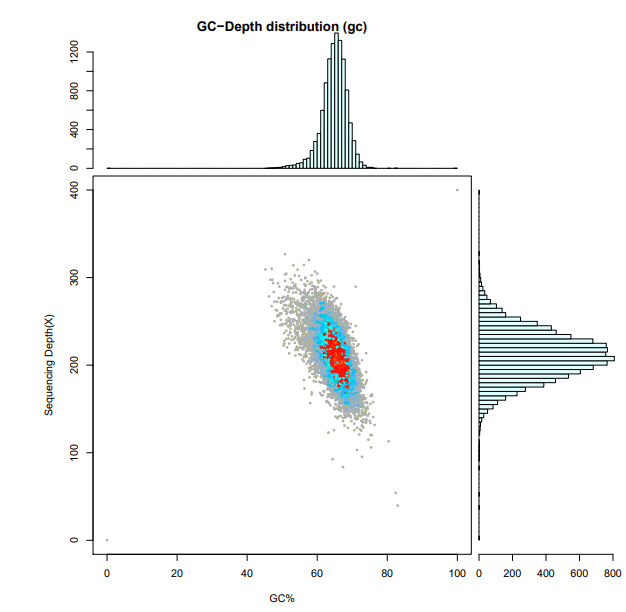
注:横坐标表示 GC 含量,纵坐标表示测序深度
3、 非编码RNA预测:使用RNAmmer 软件对基因组中rRNA进行预测;使用tRNAscan-SE 软件对基因组中tRNA进行预测。
4、 CDS预测:使用prodigal软件进行基因预测,它的目标是在识别现有基因时获得更大的敏感度,更准确地预测翻译起始点,并尽量减少错误的正向预测的数量。
Name
Value
Genome Length
6870226 bp
Number of scaffold(contig)
19
G+C content
42.7%
Num of CDS
6751
CDS num
6751
CDS total length
6139395 bp
CDS density
0.982 genes per kb
CDS average length
909. bp
Intergenetic region length
730831 bp
CDS/Genome(coding percentage)
89.4%
Intergenetic length/Genome
10.6%
GC content in gene region
43.4%
GC content in intergenetic region
36.7%
基因组信息统计
5、 COG功能注释:COG(Clusters of Orthologous Groups of proteins)是在对已完成基因组测序的物种的蛋白质序列进行相互比较的基础上构建的, COG数据库选取的物种包括各个主要的系统进化谱系。 每个COG家族至少由来自3个系统进化谱系的物种的蛋白所组成, 所以一个COG对应于一个古老的保守结构域。 构成每个COG的蛋白被假定来自于同一个祖先蛋白。 进行COG数据库比对可以对预测蛋白进行功能注释、归类以及蛋白进化分析。
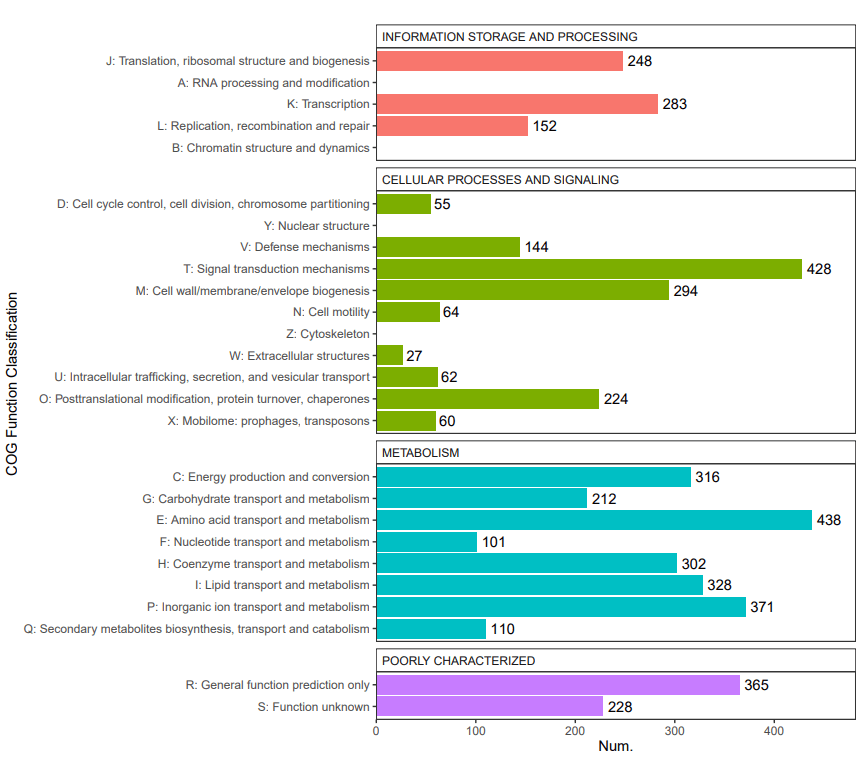 COG功能分类统计图
COG功能分类统计图
6、 KEGG功能注释: KEGG(Kyoto Encyclopedia of Genes and Genomes) 是系统分析基因功能,联系基因组信息和功能信息的大型知识库。 KEGG GENES数据库提供关于在基因组计划中发现的基因和蛋白质的序列信息; KEGG PATHWAY数据库包括各种代谢通路、合成通路、膜转运、信号传递、 细胞周期以及疾病相关通路等。
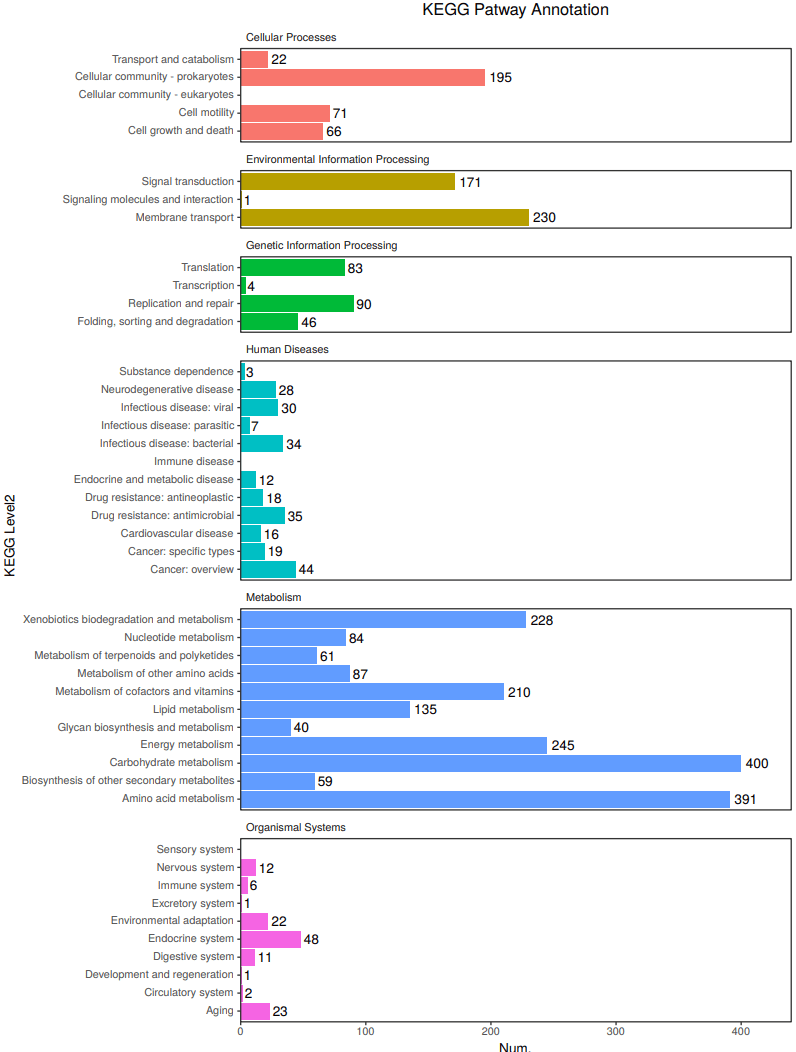 KEGG Level2 Gene Count
KEGG Level2 Gene Count
对于每张KEGG pathway图,标注基因组比对上的基因。
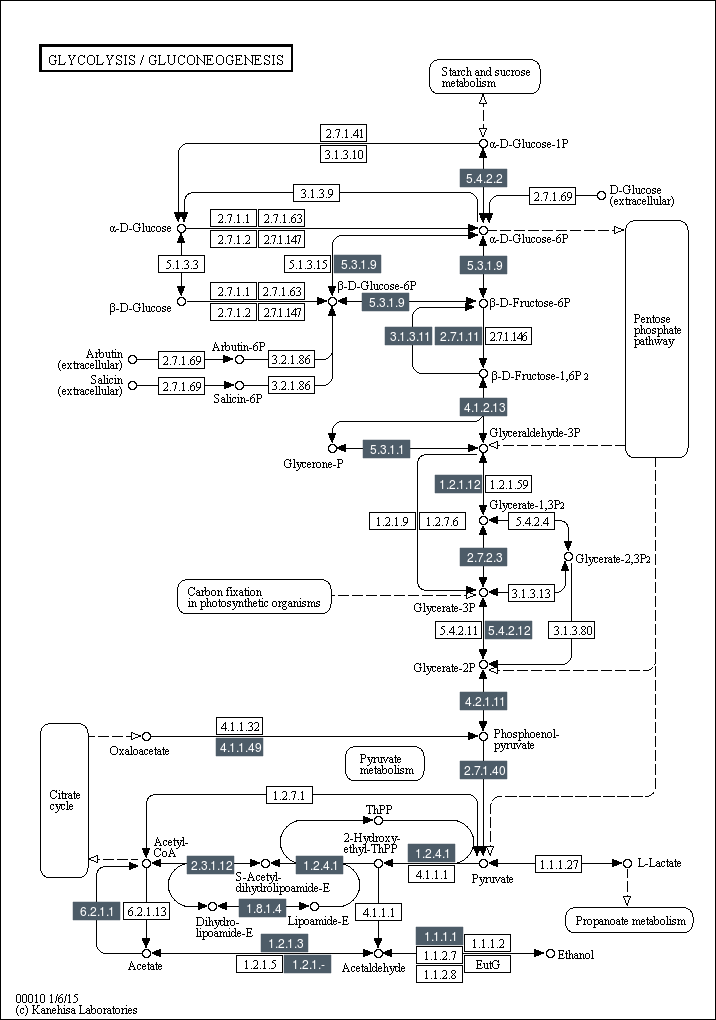 Pathway
Pathway
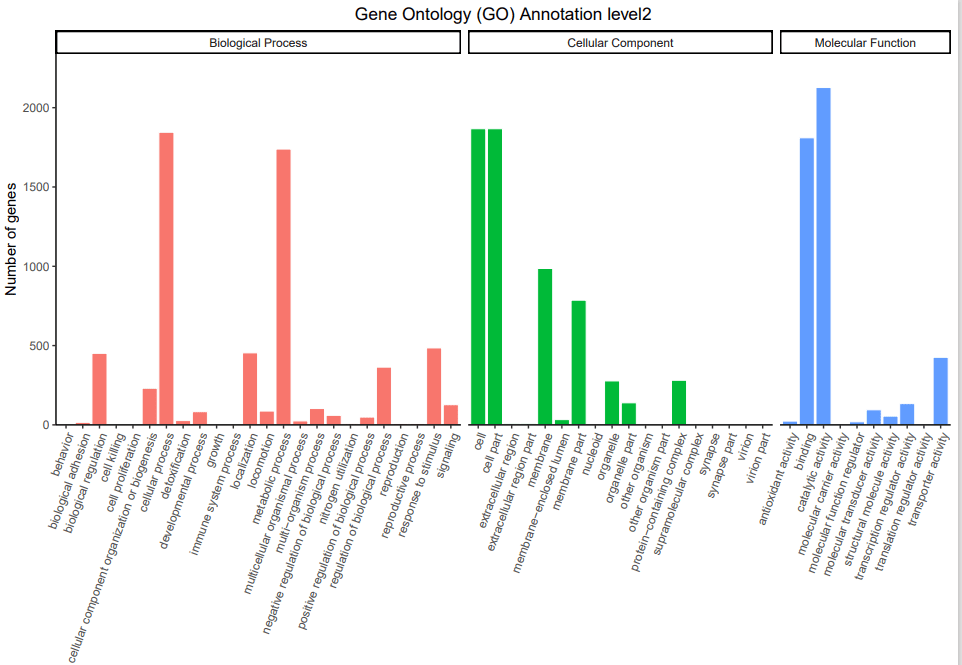
7、 GO注释: GO数据库分别从功能、参与的生物途径及细胞中的定位对基因产物进行了标准化描述,所谓的 GO,是生物学功能注释的一个标准词汇表术语(GO term),将基因的功能分为三部分: 基因执行的分子功能( Molecular Function), 基因参与的生物学过程(Biological Process), 基因所处的细胞组分( Cellular Component)。对每个分类下比对上的基因数进行统计。
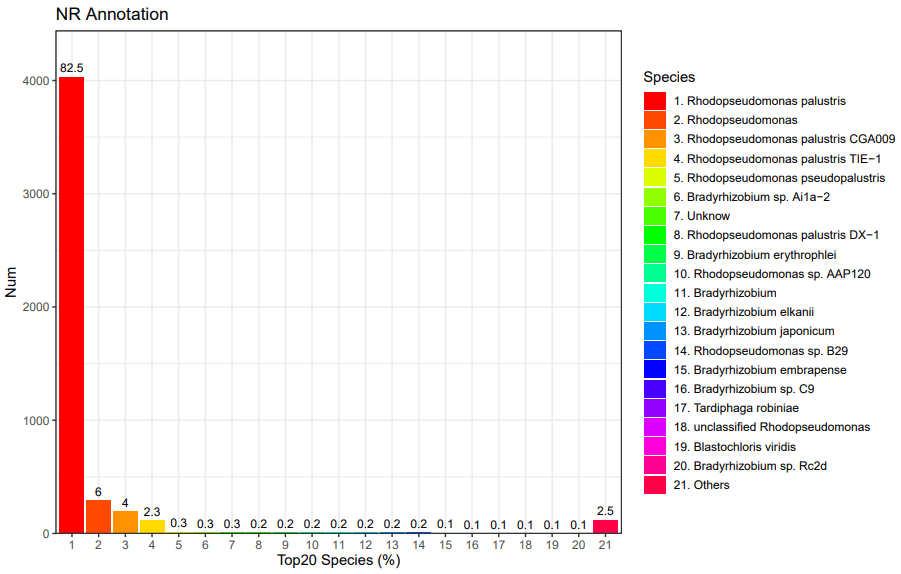
8、 NR数据库注释:NCBI非冗余蛋白数据库比对得到对应物种分类信息比率,能知道基因组物种信息。
9、 GTBD注释结果
这一分类系统以细菌中普遍存在的120个单拷贝蛋白质(bac120)为基础;在对多分组类别消歧后,根据相对演化散度标准化和分级,得到基因组分类数据库(GTDB release95)。将质控后的reads与GTDB数据库进行比对注释物种信息。
10、 Swiss-Prot 数据库注释
Swiss-Prot,是2002年由 UniProt consortium 建立的基因数据库,其特点在注释结果经过实验验证,可靠性较高,可用作其他数据的参考。
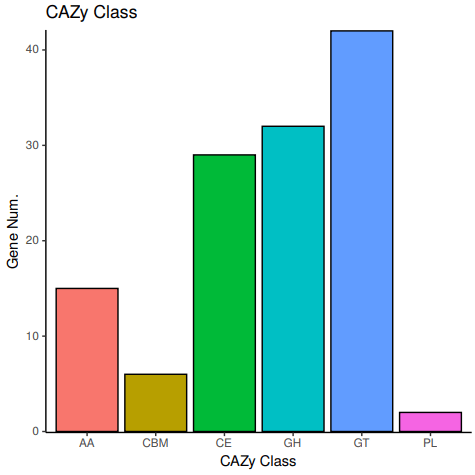
10、CAZy注释: CAZy (Carbohydrate-active enzymes,碳水化合物活性酶)数据库是研究碳水化合物酶的专业级数据库,主要涵盖 6 大功能类:糖苷水解酶(Glycoside Hydrolases ,GHs);糖基转移酶(Glycosyl Transferases,GTs);多糖裂合酶(Polysaccharide Lyases,PLs);碳水化合物酯酶(Carbohydrate Esterases,CEs);辅助氧化还原酶(Auxiliary Activities , AAs);碳水化合物结合模块(Carbohydrate-Binding Modules,CBMs)。对每个分类下比对上的基因数进行统计。
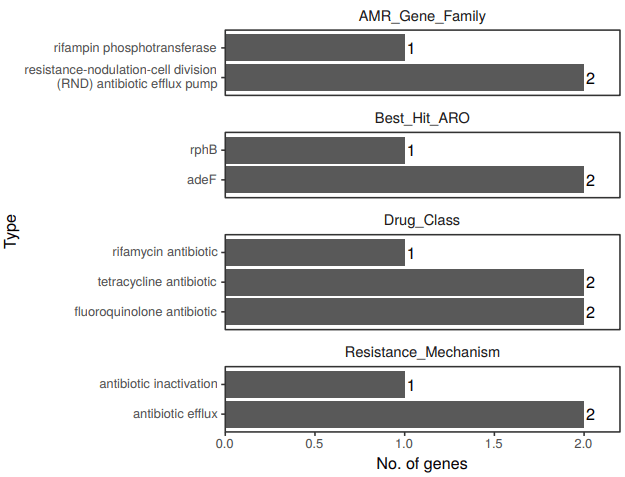
11、 CARD数据库注释: CARD(Comprehensive Antibiotic Resistance Database) 数据库,其核心是 ARO(Antibiotic Resistance Ontology),ARO 包含了与抗生素抗性基因,抗性机制,抗生素和靶相关的term。通过ARO(the Antibiotic Resistance Ontology)的形式整合了抗性基因,抗性类型,抗性机制等信息。CARD 数据库已成为目前最受欢迎的耐药基因研究工具之一。
12、 毒力因子分析VFDB
毒力因子数据库VFDB 由中国医学科学院研发,被广泛应用于毒力因子基因鉴定。
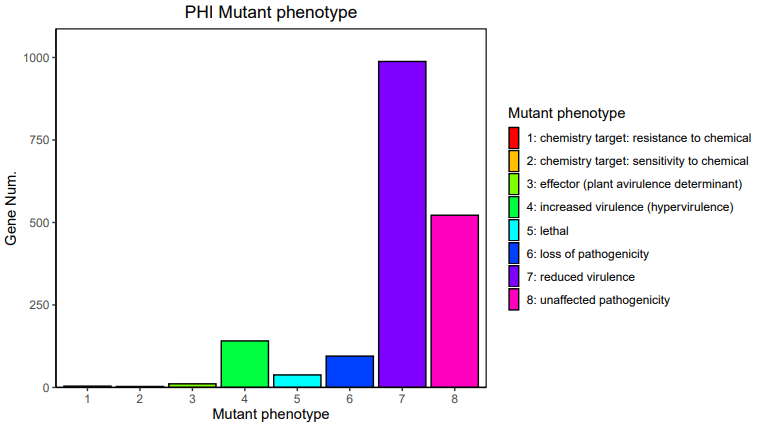
13、 PHI注释:PHI( Pathogen Host Interactions Database),病原与宿主互作数据库,主要来源于真菌、卵菌和细菌病原,感染的宿主包括动物、植物、真菌以及昆虫。该数据库对寻找药物干预的靶基因研究有重要作用,同时该数据库还包括抗真菌化合物和相应的靶基因。数据库中的每个基因都包含核酸和氨基酸序列,以及感染宿主过程中预测的蛋白功能的详细描述。病原体PHI表型突变类型基因数目的统计如下:
四、 常见问题
1、测序碱基准确率是什么意思,具体的计算方法是什么?
碱基测序的质量值Q是准确度(P)的一种格式转换,是为了方便使用一个字符表示非常复杂的准确度,占用最小空间;转换公式为P=1-10^(-Q/10),如Q=30/20/10的准确度分别为99.9%,99%,90%。
2、细菌基因组的组装结果中,N50和N90的具体含义,以及如何计算?
它们是基因组组装中常用的组装指标,要知道是越大越好。大于N50长度的序列占基因组总长的50%,大于N90长度的序列占基因组总长的90%。
具体计算方法:将所有拼接序列按照长度从大到小排列,找到TopNr 序列总长度刚好大于基因组总长度的50%(90%)位置,则该序列的长度定义为N50(N90);该数值反映了基因组50%(90%)以上的区域,都能被该数值以上长度的序列覆盖,同时体现了组装质量对于后续数据分析的质量贡献。
3、在有杂菌污染的情况下,为什么得不到好的组装结果呢?
不同物种会有非常多的同源序列,高度相似序列会对组装软件产生干扰,而软件为保证组装的准确性,只能将可疑的部分切断成不同的碎片序列。
4、如果关注的基因没有被注释出来,是什么原因呢?
1) 可能该基因在拼接时没有被成功拼接;
2) 该基因在目标基因组上可能压根不存在;
3) 在注释的数据库里还没有该基因的相关记录,所以无法被参考注释出来;
4) 研究的具体株菌中,可能根本不存在这个基因,还需要进一步确定该菌株中是否真的含有该基因。
5、草图与完成图的区别是什么?
一般细菌基因组草图是指根据二代测序结果拼接而成,中间存在gap的基因组,而与之相对的细菌完成图就是二代测序加上了三代测序,借助三代测序读长长的优势,完全没有gap的基因组。- 本文固定链接: https://maimengkong.com/kyjc/944.html
- 转载请注明: : 萌小白 2022年6月1日 于 卖萌控的博客 发表
- 百度已收录
