森言森语
下午开完组会,吃过晚饭终于得空干点别的,于是决定将之前写过的这篇关于基因家族分析的推文再检查完善一下。
检查日志2021年9月30日:黄底红字为更新内容,其他色彩为原文。
最近的一段时间是倦怠的,虽然忙,却实际上浑浑噩噩,没有长进。
不管怎么样,一个阶段的结束,总要写点东西来宣告它的结束,然后迎接新的开始。
已经很久没有跑步了,原本计划今年参加6月份的兰州马拉松和12月份的广州马拉松。
无奈兰马没有中签,后来也因多种原因兰马被推迟。但庆幸的是上半年的奔跑,不仅将体重恢复如以前的60Kg,也长高了几厘米。这是值得高兴的。
上面这段话是两个月之前写的,两个多月过去了,这段时间还是没有好好坚持跑步,只在中途跑过两三次,光阴怎么也捏不住就悄悄溜走。马上就要开题了,思绪万千。
基因家族分析简述及保姆级实操流程前言——关于基因家族分析的观点最近很长时间没有更新推文了,其实是懒惰了。今天刚好得空,就花点时间写一下我关于基因家族分析的一点粗浅理解,并整理全部的实操流程。
其实刚开始写的一些推文都很用心,也很优质,我自己也会经常返回去再看,然后再修改再完善,就像这一篇。所以还请大家见谅无规律的修订后发布之前发过的内容。毕竟以前写过的东西有些现在用的时候还是会再去回忆和完善。
学术圈的各种歧视链其实也蛮明显的,搞机理的大都觉得基因家族分析类文章都是水文,很多情况下都很鄙视基因家族类文章。但实际上没有谁的履历上全是基因家族分析类文章,基因家族分析类文章只是某个阶段的中间产物。 随着生物信息学的发展,其实会发现很多高水平论文的前期数据支持都来自生物信息学分析,而基因家族分析是生物信息学的一部分,因此,不管搞不搞生物信息学,多多少少会一点基因家族分析的工作,会大有裨益。
而我决定开始涉猎一些基因家族分析类的文章的原因也很简单,研究生阶段刚开始,我就在想一个问题,看到大家都在做这个基因,那个基因,我就想知道,最初是怎么确定某个基因的,这些基因的具体信息怎么获取。后来我知道确定某几个值得进一步研究的基因需要考虑的方面很多,需要结合大课题组的研究方向。这个当还不在我所考虑范围之内。
但有一点是明确,那就是假设已知某类基因(比如Hsp20基因家族),如何从全基因组进行家族成员的鉴定和分析。亦或者如果拟南芥中有些基因做的很好,类似的我们想找自己所研究物种中该类基因的同源基因的功能。
我现在的态度是: 关于基因家族的分析,生物大类相关方向的研究生都应该掌握,尤其研究生新生。但不能天马行空,而是应该紧密联系所在课题组的大方向。因为在进行基因家族分析的过程中,会涉及到很多的生物学概念、分子生物学基础原理以及基因组相关的知识点,而在实际分析和操作过程中,可以较为深入的理解其中蕴含的分子生物学知识。同时,在学习生物信息学的过程中,多少可以初步掌握一些数据分析和绘图制表的基础。
简单总结:通过基因家族分析可以收获哪些?
- 通过实操理解一些分子生物学实验的原理和基因组概念
- 掌握一定的数据处理基础
- 掌握基本的绘图制表方法
- 尽可能的学一些程序语言
- 结合大方向,最好能把分析的结果汇总起来简单发一篇SCI,作为小试,积累点文章写作的经历和经验
- 基因家族分析过程中学到的内容,即使没有条件单独发表文章,也一定会在将来的课题中起到很好的辅助作用
- 提高严谨性和高要求(通常在学习基因家族分析的过程中会涉及到很多细节性的问题。而在解决这些问题的过程中会逐渐提高学术严谨性和高标准)
总之,我认为基因家族分析对于研究生来说是有必要专门花点时间走一遍流程的。
好吧,废话到此为止。
下面系统的整理一下我在基因家族分析流程中的详细步骤及一些思考。
(步骤非常详细,尽可能确保看到这篇推文的读者都能完全按照步骤和流程进行分析,所以,这篇推文也会很长。)
主要内容
1 基因家族成员的鉴定进入正文(这是最关键、最重要的一步,也是我认为最有用的一个环节,需要谨慎分析,这一步出现问题,后面的分析也会有较大偏差。);
1.1 基因组文件(dna, cds, protein, gff3 or gtf注释文件)的获取;
1.2 基因家族隐马尔可夫模型(HMM model)的获取;
1.3 若没有对应的HMM model,则需要自己构建;
1.4 通过HMM model在基因组文件中搜寻 候选基因家族成员;
1.5 通过本地blast+获取参考基因家族的同源序列 (候选基因家族成员);
1.6 获取候选基因家族成员的序列信息;
1.7 结合系统发育树、保守结构域和保守基序(motif)等确定最终的基因家族成员;
1.8 检查注释文件是否正确。
2 基因家族成员序列基本信息的获取
(简单操作,实际上很多结果可能用不到,会在实验设计过程中起到一定的参考。)
2.1 基因家族成员蛋白的理化性质;
2.2 跨膜结构域、信号肽、亚细胞定位等;
3 构建系统发育树
(系统发育树是一个较大的版块,需要掌握的内容还是蛮多的。单就构建系统发育树来说,相对简单,一般也容易操作。但在树的可视化及美化编辑过程中往往并不那么容易,因为树可以呈现的内容可以非常丰富,至于如何展示,真的全在个人。这里先不展开。)
3.1 系统发育树的构建
3.1.1 基于家族成员全部序列的常规系统发育树的构建;
3.1.2 其他保守domain的系统发育树构建;
3.1.3 建树序列的修剪;
3.1.4 建树策略的选择。
3.2 系统发育树的美化和编辑
3.2.1 可视化进化树;
3.2.2 iTol进化树美化配置文件的准备;
3.2.3 系统发育树和其他数据(如转录组数据等)的结合。
4 转录组数据分析(基因表达量分析)
(这一部分的结果其实也只是用来筛选感兴趣基因的参考,不能作为绝对参考。也就是说不能把宝只压在转录组数据上面。 通常现在的转录组测序服务,都会附带做一些数据分析,多数情况下公司会返回logFC和tpm值,一般直接使用即可。)
4.1 基因家族成员在不同处理下的转录组数据或者定量数据的可视化;
4.2 差异基因的筛选及绘图;
4.3 表达模式相似的基因筛选(Venn图);
4.4 获取公开的已发表的转录组数据进行家族成员的表达分析。
5 其它
(其它主要包括上述没有提及的基因家族分析流程中的内容,列在其它里面,主要我个人认为真的用途不大,如果自己的基因涉及到相关的内容,可以单独进行分析。)
5.1 顺式作用元件的预测和分析
5.2 基因在染色体的定位
5.3.1 物种内共线性分析
5.3.2 物种间共线性分析
5.4 GO富集分析
5.5 KEGG分析
软件安装
所有提到的软件及安装方法都列在了文章最后。
为了编辑方便,图文可能不甚相符,因为完全从头来一遍实在需要花费很多时间,所以提示大家以文字为主,图片作为参考。(下面的每一小节内容相对是完整的,可以参考,全文的数据并不连续,需要当心,下文的十分具体的步骤仅仅是参考之一。)
1 基因家族成员的鉴定 1.1 小麦基因组的获取首先需要确定一个选题,这是最费劲的一部分,往往有一个好的idea几乎就决定了后续的直接结果导向 (因为我们的目的不只是随便搞一篇基因家族分析的文章,而是希望通过该分析能够在后续深入研究其功能,但如果仅仅是前期学习,可以选择已发表的论文作为分析对象,这样也可以检验自己的分析结果是否符合既定结果。)。不论是基因家族分析,还是其他的课题,想定题,都不容易。需要建立在大量阅读文献并和导师充分交流的基础上 (分析对象必须和课题组的发展贴合,并充分和导师沟通。否则根本无法在后续深入推进。)。
大家可以以 MIKC-type MADS-box( Triticum aestivum )基因家族为分析对象。
尝试重复2020年发表在 new pyhtologist上的文章 Genome-wide analysis of MIKC-type MADS-box genes in wheat: pervasive duplications, functional conservation and putative neofunctionalization 的部分结果。
首先,不管到底MADS-box是个什么情况,先将目标物种(小麦)的相关基因组数据和注释文件下载下来,推荐使用EnsemblPlants数据库 http://plants.ensembl.org/info/data/ftp/index.html,这个数据库包含了大量的物种基因组信息,除了可以下载小麦基因组之外,大多数物种的基因组都可以从这里获取, 主要是该网站包含相对完整的注释信息。当然,个别新发表的物种基因组可能还未收录其中,需要查阅相关文献进行获取,一般发表的基因组信息都会挂在网站上面(基因组文件动辄几个G,推荐IDM下载神器进行下载)
比如搜索小麦拉丁文( Triticum aestivum ),可以将 DNA , cDNA , CDS , Protein sequence , Gene sets(GTF&GFF3) 这几个文件都下载下来,以后会用到。其他的文件大家就自行探索吧。下载好之后在后续分析过程中直接使用就好,不建议进行手动修改,尤其不建议对注释文件(GTF,GFF3)进行修改。
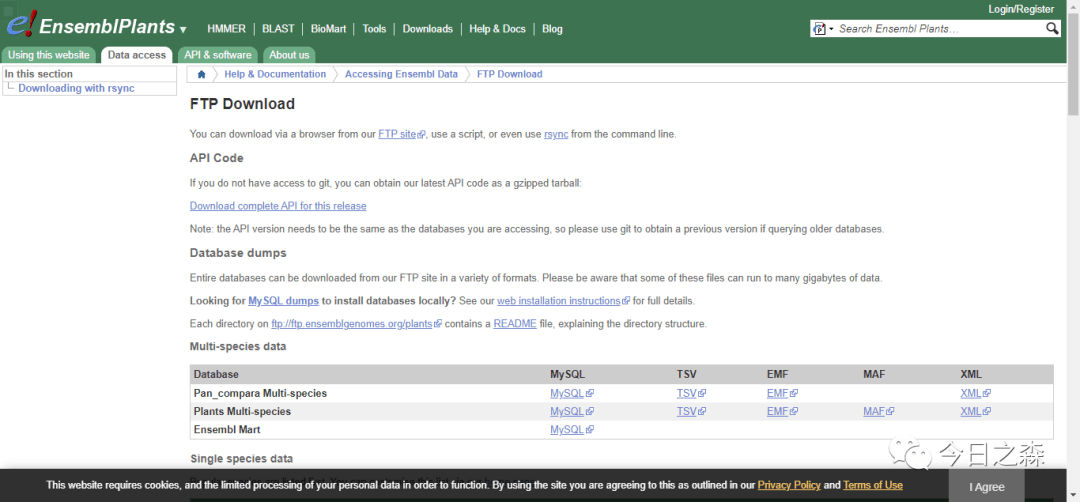

下载好之后需要先进行简单操作,然后备用。
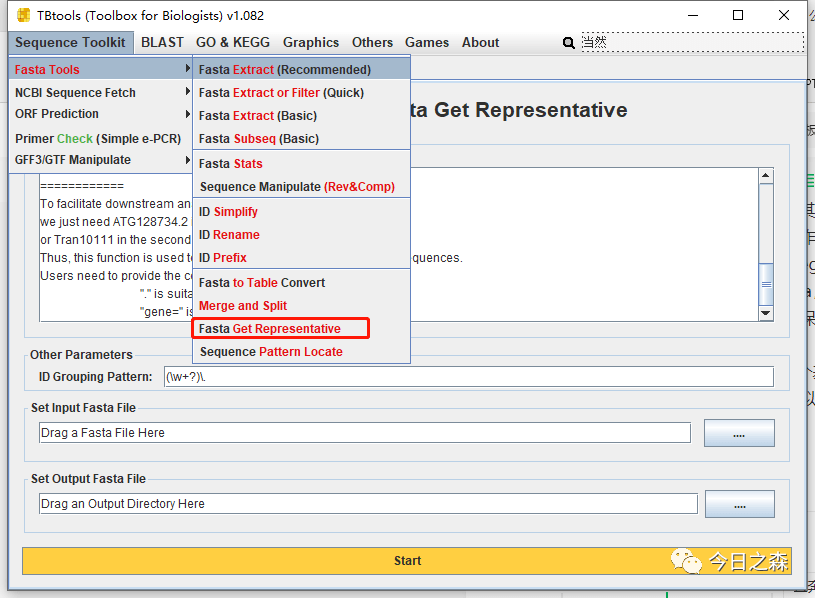
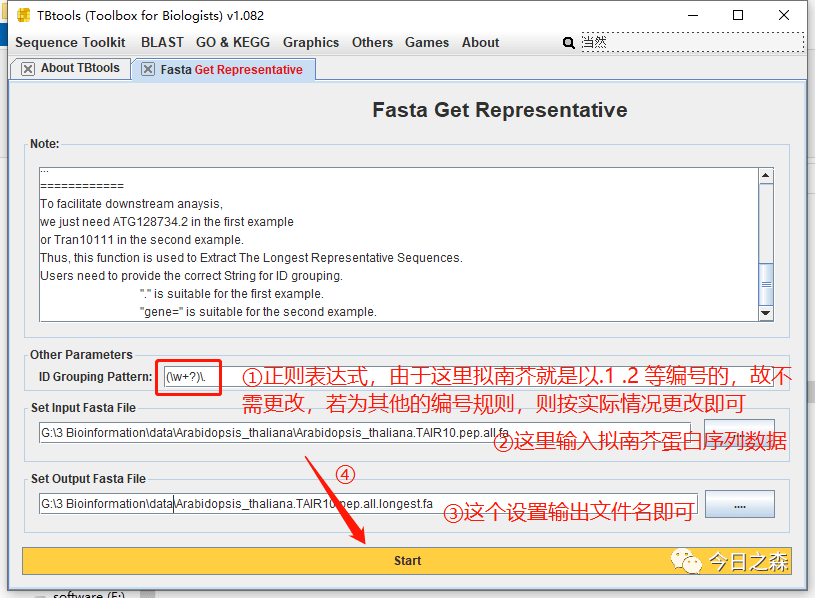
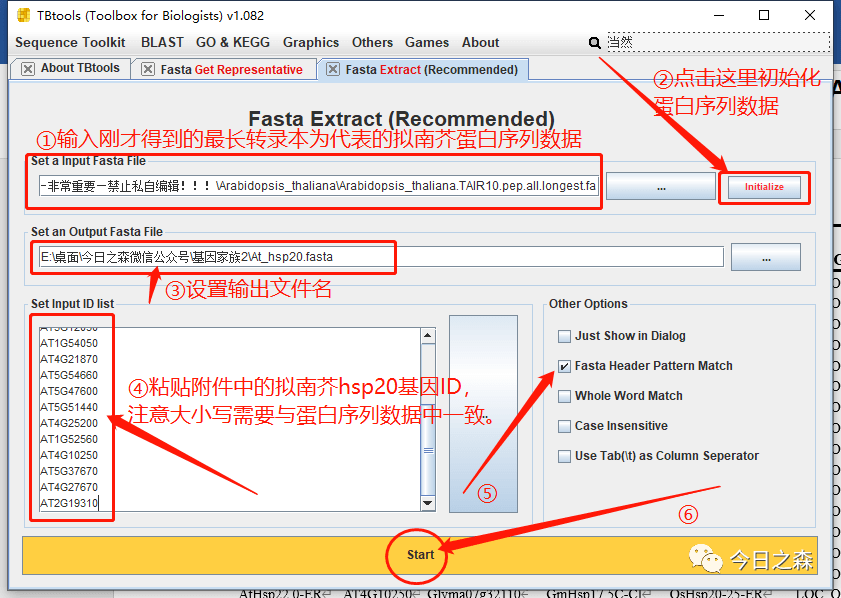
基因组文件中通常同一个基因会有多种长度不同的转录本,在进行基因家族分析时,我们经常默认使用最长的转录本作为一个基因的代表序列。这样在后续的分析中,一个基因对应一个转录本。
所以,这里需要提前对下载好的基因组文件中的 CDS 和 protein 文件进行处理,获取最长转录本。
如果所研究物种的基因组注释文件gff文件不是很完整,也可以使用tbtools进行核验校准。这里没有标出,可以参考微信公众号《生信药丸》。
这篇2020文章中下载的是1.0版本的小麦基因组文件及相对应的注释文件。我们这里选择下载组装质量较高的1.1版本的小麦基因组文件及对应的注释文件。
注:有的物种基因组有多种版本,需要获取组装质量相对最高的版本进行下载。这时候需要格外仔细,最新版的基因组不一定组装质量就最好,即使被公开发表或已收录进相对权威的数据库,也要仔细筛选,一般这种情况较少。
1.2 隐马尔可夫模型文件的下载比如“中国春”小麦基因组信息自2014年IWGSC发表第一版之后,产生了7个版本。
- RefSeq v1.0版本:TraesCS1A01G000100.1 下载地址:https://wheat-urgi.versailles.inra.fr/Seq-Repository/Annotations
- RefSeq v1.1版本:TraesCS1A02G000100.1 下载地址:https://wheat-urgi.versailles.inra.fr/Seq-Repository/Annotations
- RefSeq v2.0版本:(2.0基因组已经公布,注释基因声称2020年1月发布,但至今好像还没有看到2.0版本的注释信息,这次最新的版本为v2.1,这应该是也是为了有别于2.0而重新定义的版本号) 下载地址:https://wheat-urgi.versailles.inra.fr/Seq-Repository/Assemblies/v2.0/
- RefSeq v1.2版本:(这是一个过渡版本,对RefSeq v1.1版本的局部进行了完善。) 下载地址:https://urgi.versailles.inrae.fr/download/iwgsc/IWGSC_RefSeq_Annotations/v1.2/
- RefSeq v2.1版本:TraesCS1A03G0000200.1(目前最新的“中国春”参考基因组,但其组装质量却非最高,感兴趣的朋友可以邮箱联系我进行交流。) 下载地址:https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Annotations/v2.1/
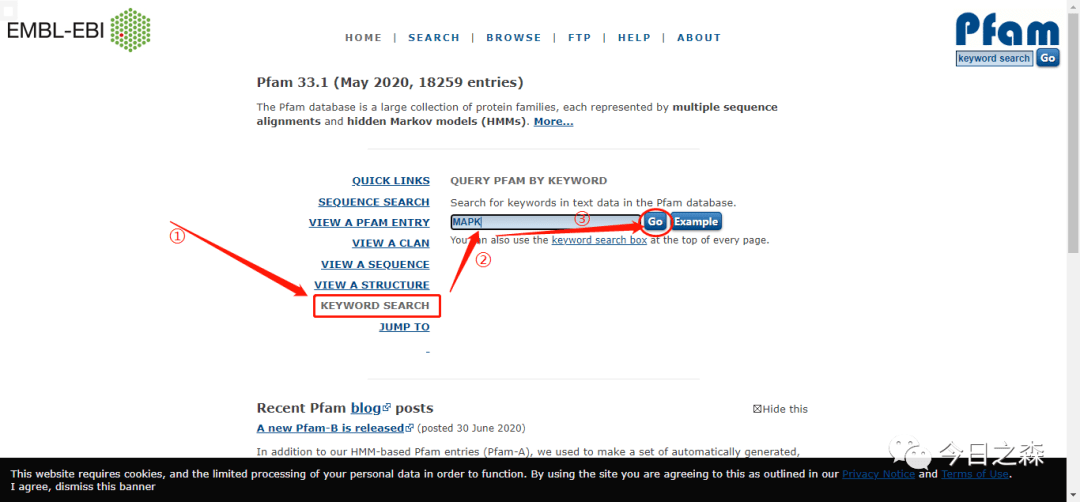
从文章得知,MADS-box基因家族包含两个保守结构域 MADS(PF00319) 和 K(PF01486) 。
在Pfam网站https://pfam.xfam.org/下载隐马尔可夫模型文件,文中已经给出了具体的Pfam号,其实也可以直接搜索保守结构域名称。
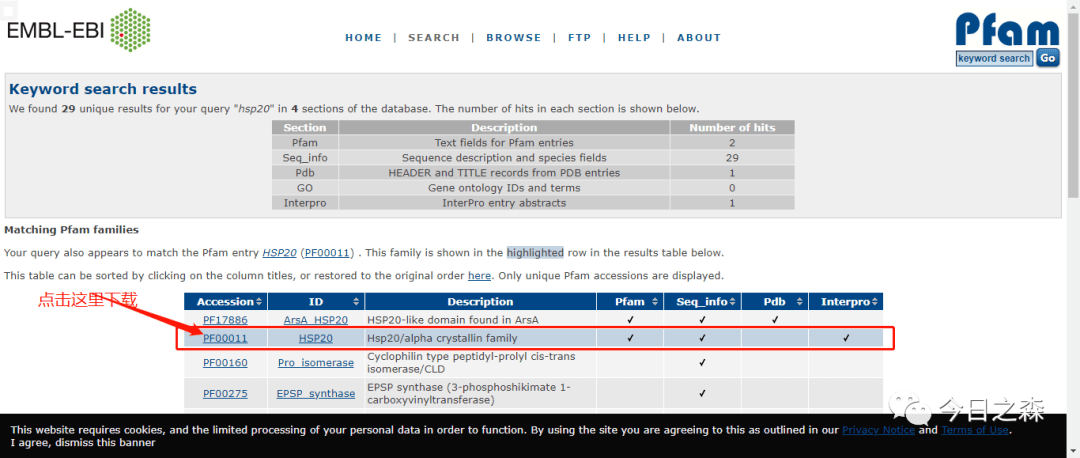
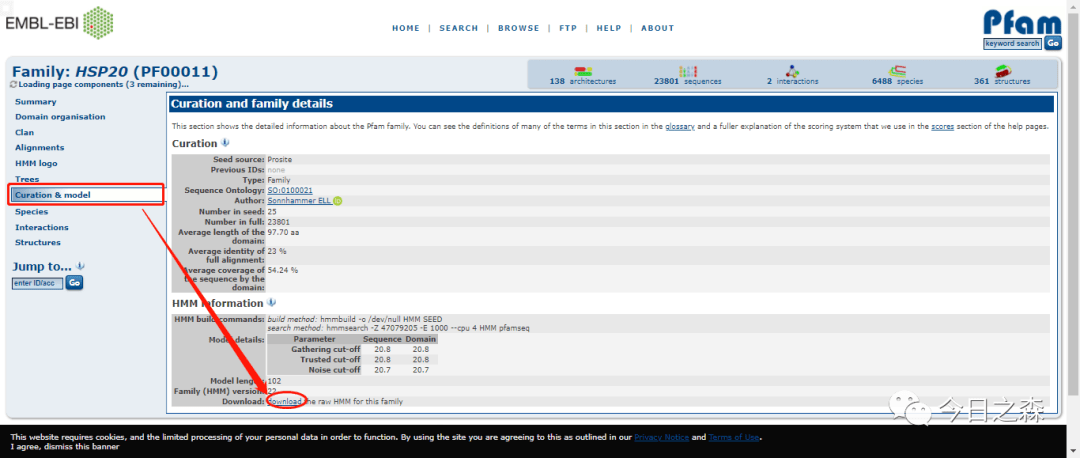
1.3 如果没有对应的HMM model文件,需要自己构建
上述我们选择的保守结构域有对应的HMM model文件,可以直接在Pfam网站下载,如果没有,就需要自己构建,也相对简单。那我们假设没有MADS-box的隐马尔可夫模型。
主要的构建逻辑就是直接在NCBI网站搜索MADS-box,肯定会出现很多结果,为了我们我们构建的HMM model相对精确,可以在搜索时指定几个物种,比如指定拟南芥和水稻作为搜索物种,这时会搜索到所有与拟南芥和水稻相关的MADS-box的基因,将搜索到的所有基因的蛋白序列保存为fasta文件。然后以这个文件为背景构建MADS-box的HMM model文件。
当然,这样其实也不是很好,最好的方法是,想办法获取到拟南芥和水稻已经发表的MADS-box蛋白序列,这样就可以构建出更为准确的HMM model文件。最好是这样,不过也不影响。
#align .fasta是多序列比对结果文件, MADS-box .hmm是自己命名的 HMM model文件构建HMM model的流程
1.将上述搜索到的全部基因的蛋白序列首先进行多序列比对,得到align.fasta文件,多序列比对这里可以用MEGA软件中的MUSCLE,但我更推荐mafft软件进行多序列比对,因为文中总共鉴定了439个MADS-box基因家族成员,如果使用MEGA比对,会很慢,经常容易出现卡死的情况,而且mafft不仅在比对速度和比对质量上,都是目前最优的选择之一,后续我们所有的多序列比对都用mafft;
2.然后通过下述命令行操作,即可得到HMM model。
hmmbuildalign.fastaMADS-box.hmm
这一部分先不写了,大家有需要的可以发邮箱咨询,或再今日之森学术交流群和大家交流讨论。1.4 利用HMM model在基因组文件中搜寻候选基因家族成员
实际上前面提到了一句,获取候选基因家族成员有两个策略。
一是用下载好的hmm模型或自己构建的HMM moedl模型作为query,来搜索马铃薯蛋白序列数据。在这里就需要用到hmmer子程序 hmmsearch 。
二是用已发表的确定的MADS-box序列为种子序列(query),直接本地blast获取同源基因。
第一种策略
MADS-box有两个保守结构域HMM model文件,这时,我们可以分别使用PF00319和PF01486去search小麦基因组,如果分别search,需要操作两次,最后还需要将得到的结果进行汇总,操作起来相对不爽。所以我们可以提前手动将上述两个HMM model文件进行合并,合并为一个HMM model文件,就像操作txt记事本那样,把其中一个复制后粘贴在另一个后面,然后重命名即可,比如这里我们重命名为MADS-box.hmm。然后操作下列代码即可得到search结果。
hmmsearchMADS-box.hmmTriticum_aestivum.IWGSC.pep.alllongest.fa> MADS-box.out
第二种策略
学会本地blast其实蛮有用的,通过blast获取候选基因家族成员的原理比较好理解,实际上都是获取参考物种(比如拟南芥和水稻)基因家族的同源基因。
运行以下代码即可。
#首先以小麦全基因组蛋白质序列构建数据库
makeblastdb-inTriticum_aestivum.IWGSC.pep.alllongest.fa-dbtypeprot-outTa
#然后进行本地 blastp
blastp-queryMADS-box.fasta-dbTa-evalue1 e-5-num_threads4 -max_target_seqs10 -outMADS_box_blast_pep.txt-outfmt6
1.6 获取候选基因家族成员的信息
通过前面1.4和1.5两步,我们得到了hmmsearch和blastp的结果,前面在1.3页提到了hmm构建时的不影响。
在这里就可以更好的理解,为什么前面自己构建隐马尔可夫模型时,指定物种与否影响不大。
因为到这一步位置,我们得到的基因家族成员都统称为候选基因家族,之所以称为候选基因家族成员,顾名思义,是因为还没有最终确定下来,所以我们在:
- 构建hmm model时,如果不指定物种,得到的序列就很多,构建的hmm模型文件也相对丰富和庞杂,其中免不了出现很多冗杂的信息,hmmsearch得到的候选基因家族成员也就更多一些;如果制定了物种,得到的序列就相对单一,构建的hmm模型文件也相对可靠,最后hmmsearch得到的候选基因家族成员也就更少一些。但不论多与少,我们都不能轻易通过阈值等剔除出去。
所以我们在进行基因家族分析时,往往需要在这一步之前尽量放宽限制条件,让尽可能多的序列包含在我们要分析的基因家族当中。
因此,这一步,我们需要获取得到前述全部候选基因家族成员的序列信息,主要是 protein 和 CDS 序列。
通过gene ID即可获取。
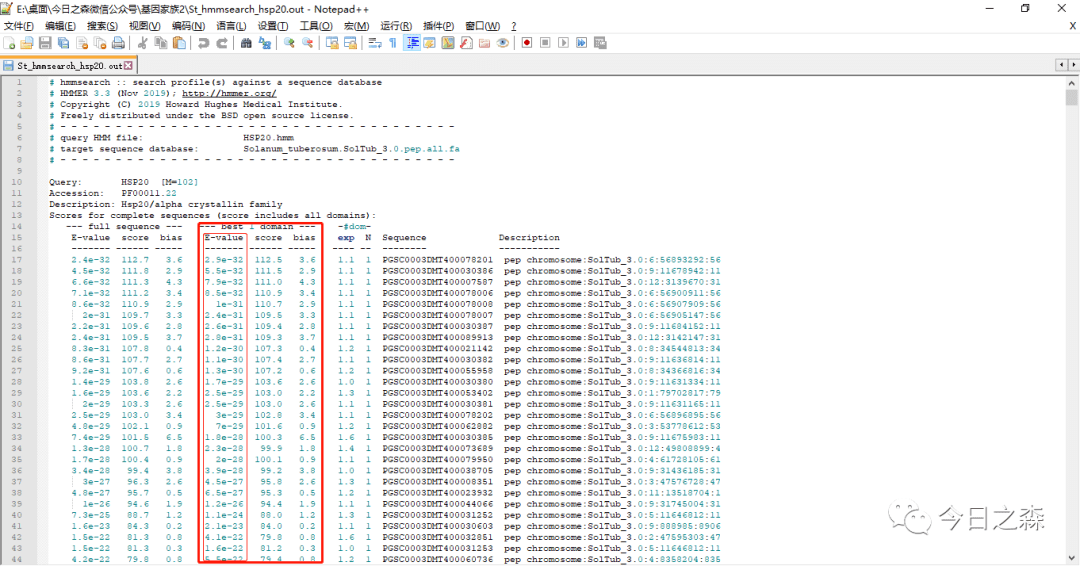
这里展示了几列信息,很好辨识,左边三列是全序列比对结果(按score从高到低排序),红框中的三列是hmm模型结构域最佳匹配结果(排序同上)。
这里我们主要以红色框中的搜索结果为准,由于搜索到的序列较少,故不以e值限制(一般以10-5进行限制),即将搜索到的结果全部保留。
如果要blast
输出文件即为比对结果。打开后如下:
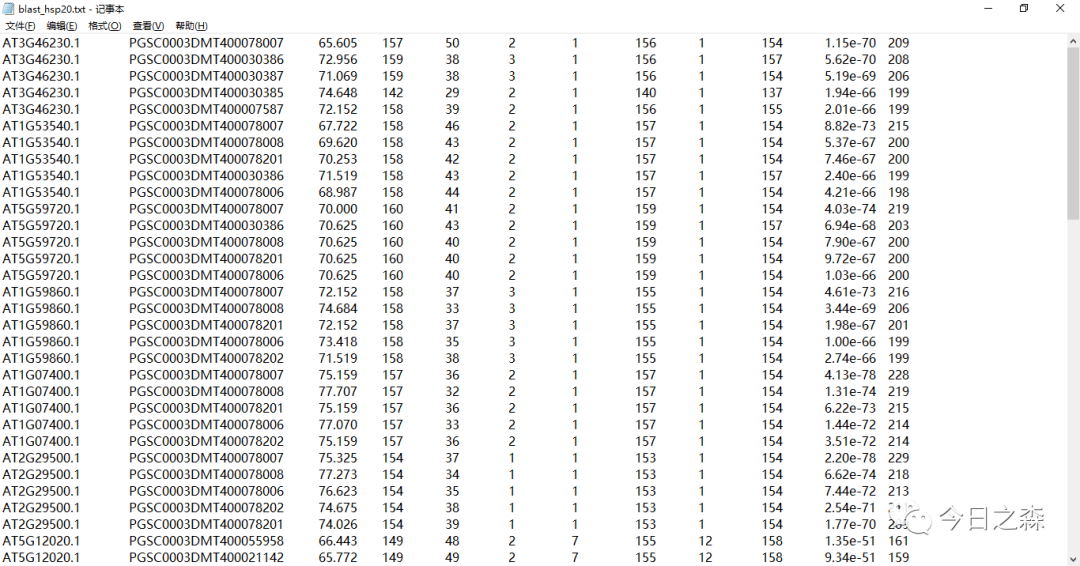
有时候为了结果更加准确,可以把hmmsearch结果和blastp结果合并。
将两种方式获得的基因进行合并,去掉重复值,注意,这里仅仅是去掉重复值,并不意味着去掉重复值后就是马铃薯hsp20基因家族的成员,因为,去掉重复值后还需要进一步研究每个基因所包含的结构域,这就需要明确hsp20基因包含哪些保守domain。

注意看,得到的基因ID后面有很多描述信息,这是我们不需要的,因此还需要借助tbtools进行ID简化。
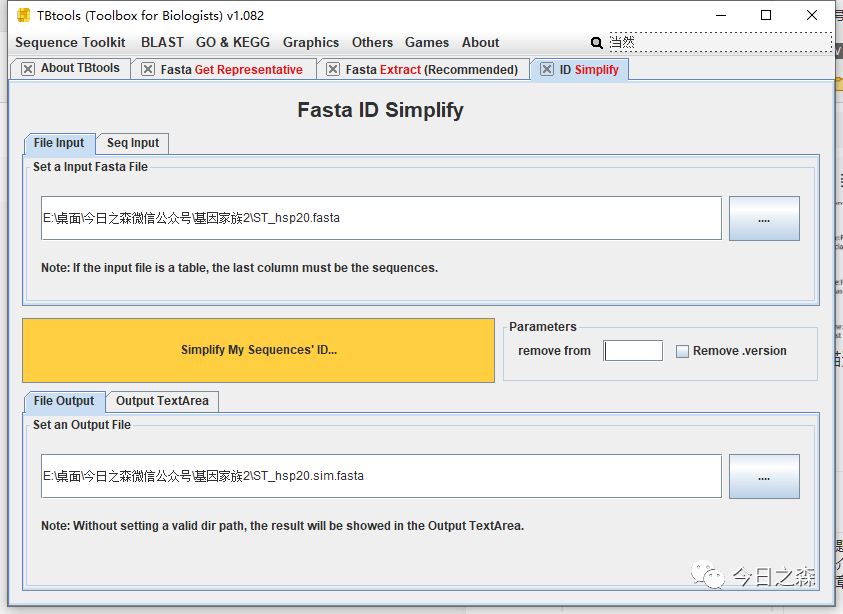
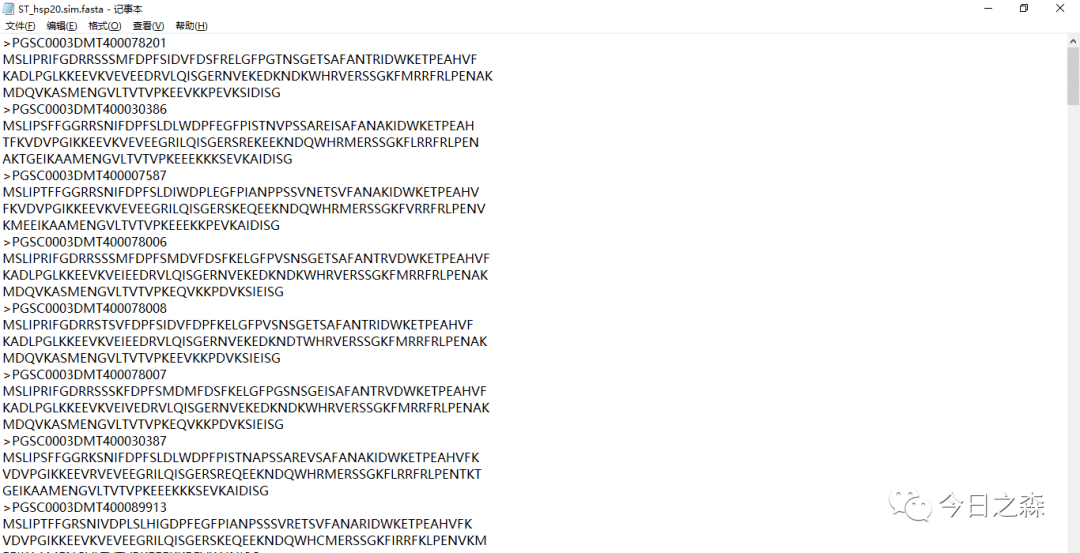
1.7 结合系统发育树、保守结构域和保守基序(motif)等确定最终的基因家族成员
这一步是最关键的一步,因为涉及到剔除候选基因家族成员中不属于该家族的成员。
一个是考虑构建系统发育树,然后查看保守结构域是否完整,有没有明显的缺失等情况。
文献中表明,MADS-box基因家族的成员,可以同时含有 MADS(PF00319) 和 K(PF01486) ,或者仅包含其中一个结构域均被视为MADS-box家族成员。
1.8 检查注释文件是否正确
这是什么意思呢?在1.7中我们决定剔除某些结构域明显不完整的基因时,往往不能太草率,因为剔除出去,意味着后续我们将永远不再考虑它了,所以在剔除这一步应该相对谨慎,起码要严谨。
因为有的时候某个基因出现结构域残缺不全的情况,并非该基因不属于我们要研究的基因家族,而有可能是注释文件不完整造成的。
这种情况常见但又不常见,好吧,这是一句废话。
总之,如果我们研究的候选基因家族成员中偶尔有一两个类似的基因,那我们就有必要检查注释文件是否准确,如果准确,该基因缺失残缺不全,那我们就认定为它是一个假基因,可以将其提出出局;如果通过一番验证,发现其缺失是注释文件搞错了,那我们有必要矫正注释文件,给这个基因重新注释一下,然后保留。
小结:到此为止,我们就确定了MADS-box家族成员,得以进入后续的分析。
2 基因家族成员序列基本信息的获取
这一部分,就是对我们确定的基因家族的信息进行简单汇总,通常我们会形成一个表格。包含如下信息:
gene ID|subfamily|CDS length|protein length|Molecular weight|等电点|CDS sequence|protein sequence|
- 蛋白物理化学性质分析 https://web.expasy.org/protparam/
- 亚细胞定位 http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/#
- 跨膜结构域预测 http://www.cbs.dtu.dk/services/TMHMM/
- 信号肽预测 http://www.cbs.dtu.dk/services/SignalP/
这里只是提供了一些我常用的网站,其实现在每年都会有很多类似的网站发表,有时甚至会有针对某个物种的预测网站。操作起来相对简单,将蛋白fasta文件提交后等待片刻,即可获取到对应的信息。然后稍微花点时间整理为表格即可。
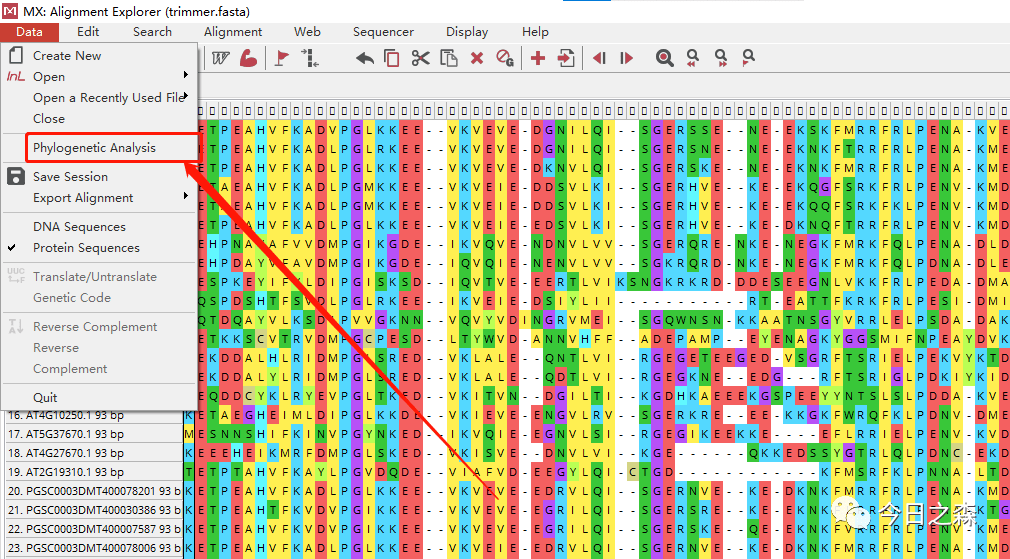
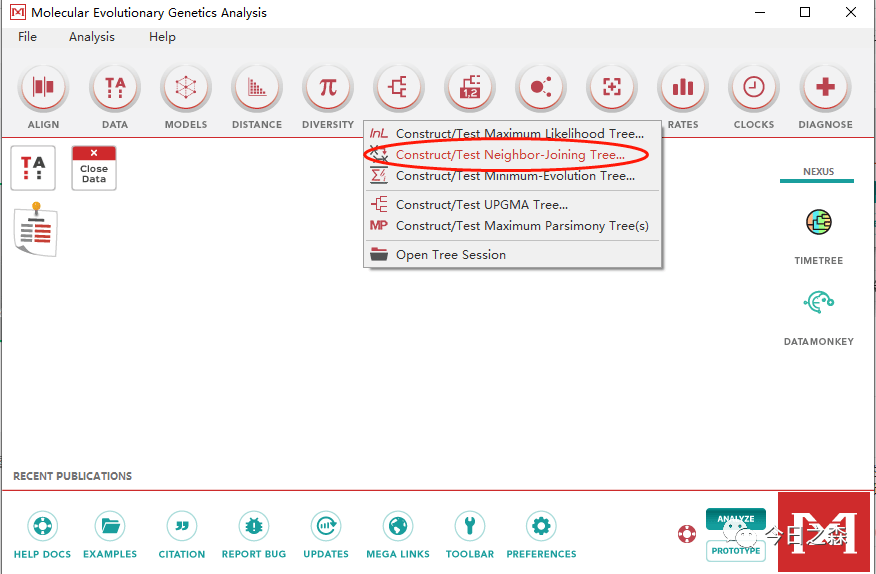
3 构建系统发育树
3.1 MEGA构建系统发育树
利用MEGA构建系统发育树,只需要准备前述确定的基因家族成员蛋白质fasta文件节课,导入MEGA后完成比对和建树,并导出树。
但是我们这里序列较多,同样采用 mafft 进行多序列比对,然后利用 fasttree 建树,最后用 iTOL 美化树。
这里大家可以直接看我以前的推文:
基因家族分析(1)——进化树构建与美化
进化树美化:iTOL常用配置文件的使用,你一定需要!
关于多序列比对,这里只介绍两个软件,一是MEGA,虽然该软件操作简单,也集成了Clustalw和Muscle(如下图),但适合配置一般的电脑进行少于500条以内的多序列比对(经验之谈);二是mafft,需要基于命令行操作,但tbtools中好像也集成了mafft,感兴趣的可以去研究,mafft的最大优势是可以快速进行几千条序列的比对,是我用过最快的多序列比对软件,当然还有其他优势。这里有几种情况
- 对于常规建树,多序列比对后直接建树即可;
- 对于基因家族庞大(几百几千,但又包含唯一保守结构域)的情况下,可以提取保守结构域序列后,对保守结构域序列进行多序列比对并建树。
- 建树后往往需要一定的修剪,但非必要。也就是说多序列比对后可以选择对gab进行删除,或者手动将两边比对不是很好的区域直接删除,但不管怎样,这样是比较随意的。所以我一般要不就是直接拿多序列比对结果建树,要不就是使用 triml 修剪。 我没有人为的删除过两端的序列。
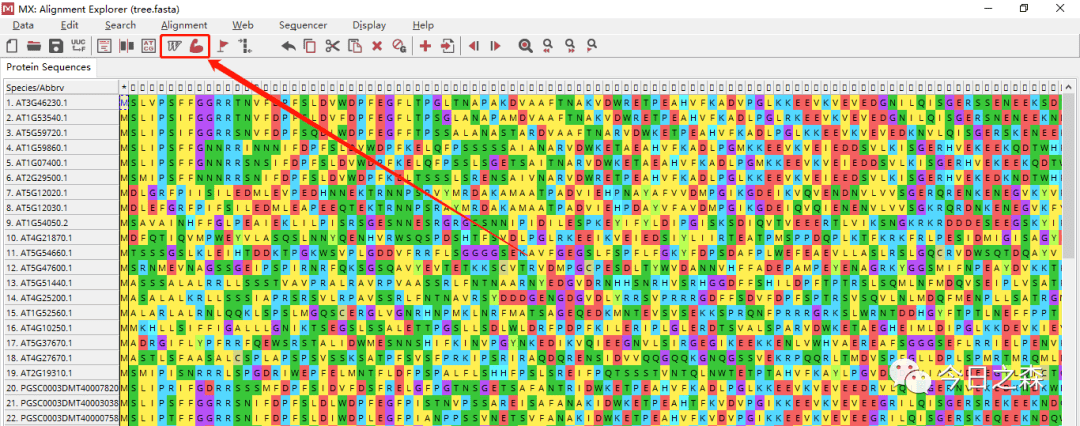
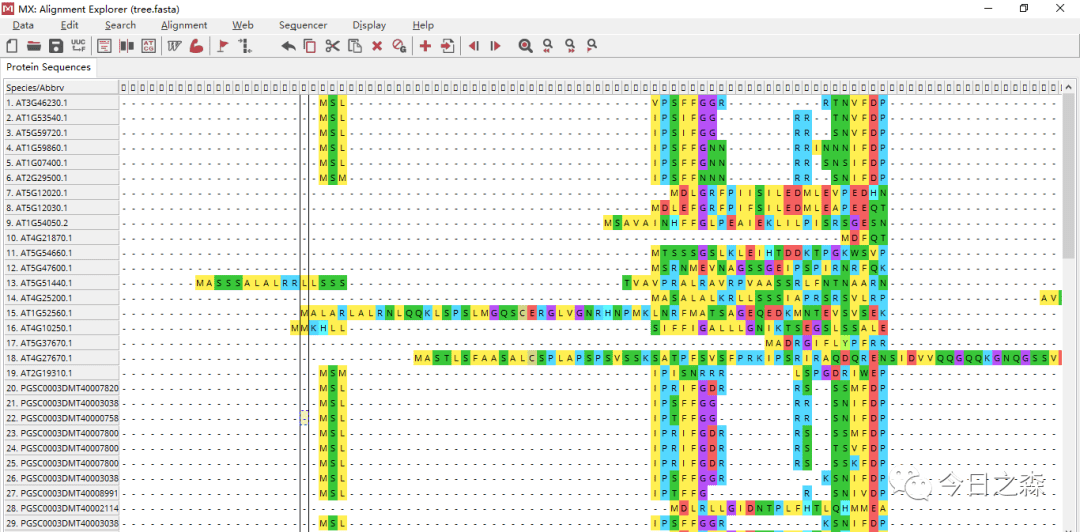
从这个比对结果来看,差异是非常之大,由于序列较少,继续建树应该影响不大。
可以看出,这样的结果使我们不想要的,但是由于序列较少,基本不影响建树的准确性。但是为了进一步。我们需要进行修剪多序列比对结果,以构建更准确的进化树。
先讲MEGA多序列比对结果导出为fasta格式。然后打开tbtools,使用Trimmer快速修剪。
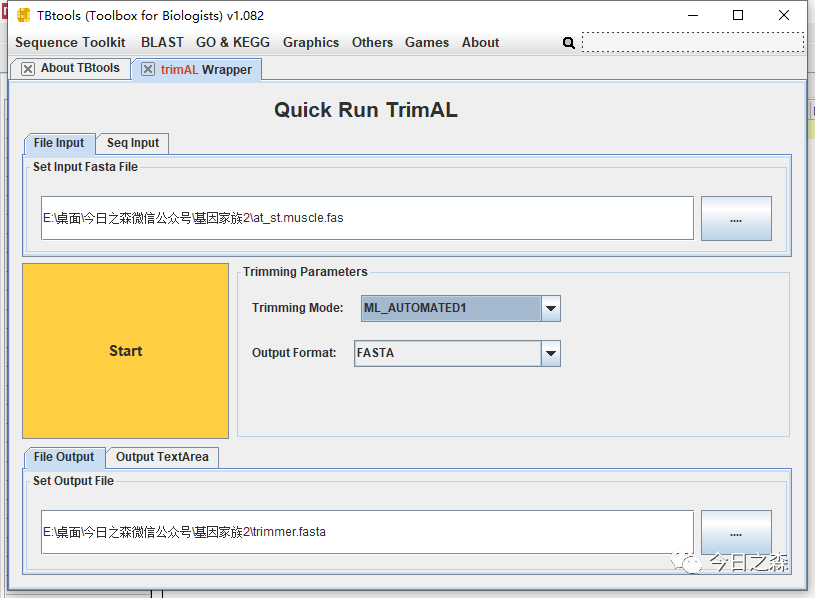
修剪之后,就美好了很多,如下:
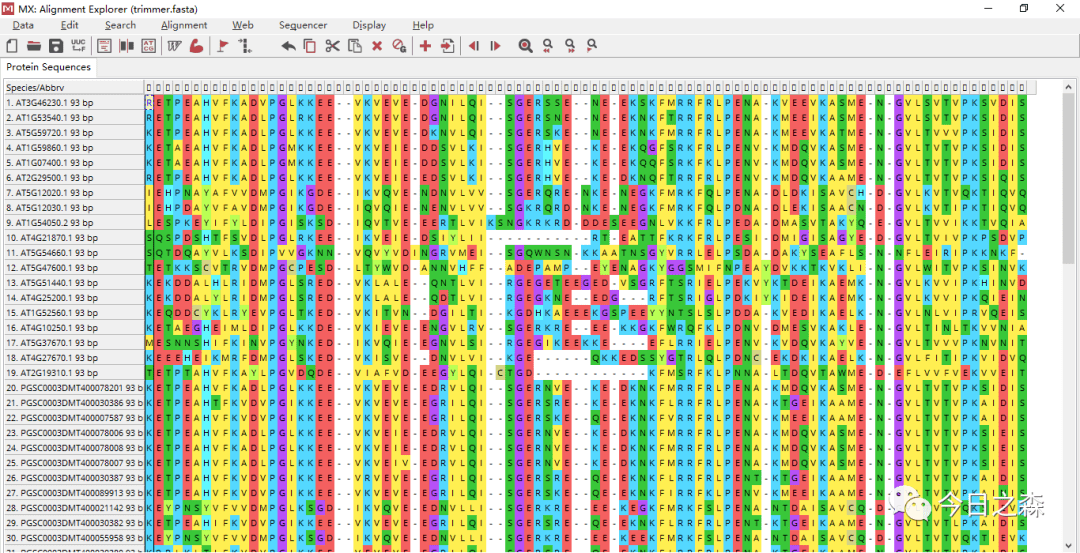
进化树构建
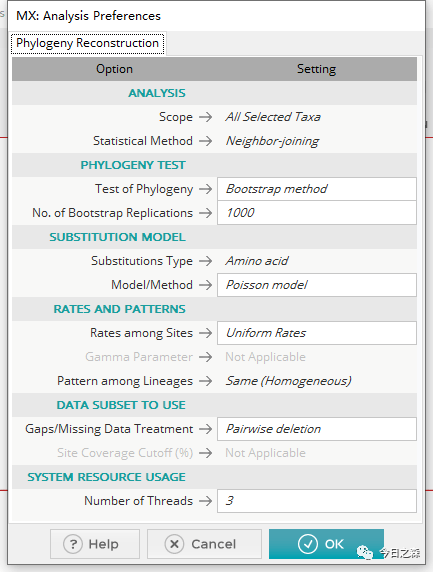

导出后生成以.nwk结尾的树文件,这个文件可以用其他进化树美化软件打开后进行操作。
4 转录组数据分析
这一部分怎么说呢,现在公司测转录组之后,一般都会附带进行转录组数据分析,也就是从 count 到 tpm ,一般情况下我们只需要对这个结果利用不用的阈值进行筛选即可。
而且,通常还会进行GO和KEGG注释。一般来说不需要我们额外做什么。因为前段时间我通过R-Package “ edgR ”和“ DEseq ”验证公司的转录组数据分析结果,发现一模一样,那我还费啥劲呢!
但是这里还是以部分公开发表的转录组数据进行简单分析。
5 其他
在目录中已经提及,我要在第5章其它中提及的内容,咋说呢?这些内容在基因家族分析的常规文章中都有提及,但是实际上有啥用处,我真的不能理解。实话说,我感觉顺式作用元件,GO注释,KEGG之类的,不管啥基因,最后结构都差不多,如果不是的话,一定是我孤陋寡闻。不管了,反正,别人有的,我们也必须要有。那就接着往下走。
5.1 顺式作用原件的预测和分析5.2 基因在染色体的定位5.3.1 物种内共线性分析
总体来说,需要以下三个文件,就可以实现物种内共线性圈图。
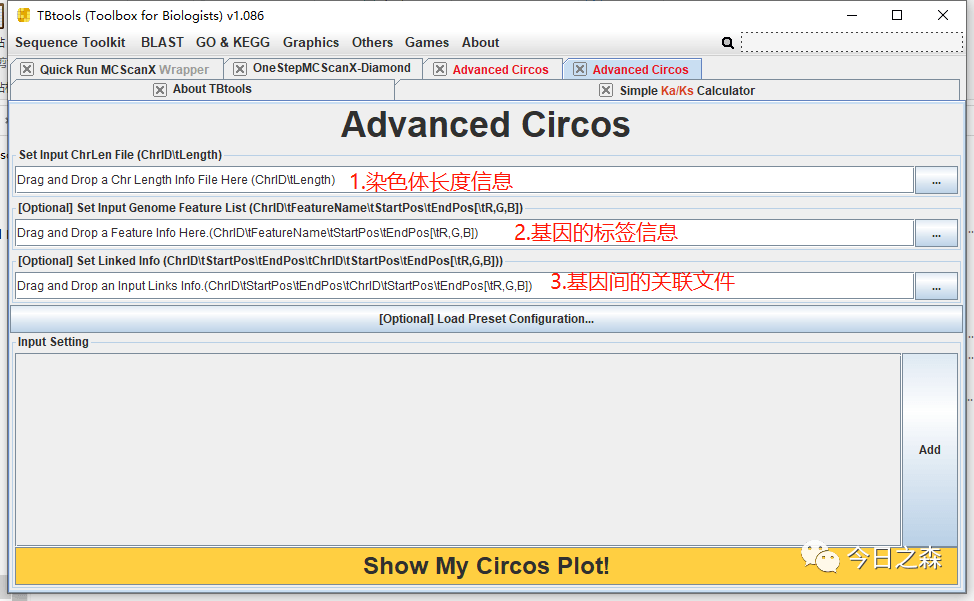
第一步:先准备染色体长度信息
首先打开tbtools,进入Fasta Stats。按照下列示例输入文件。
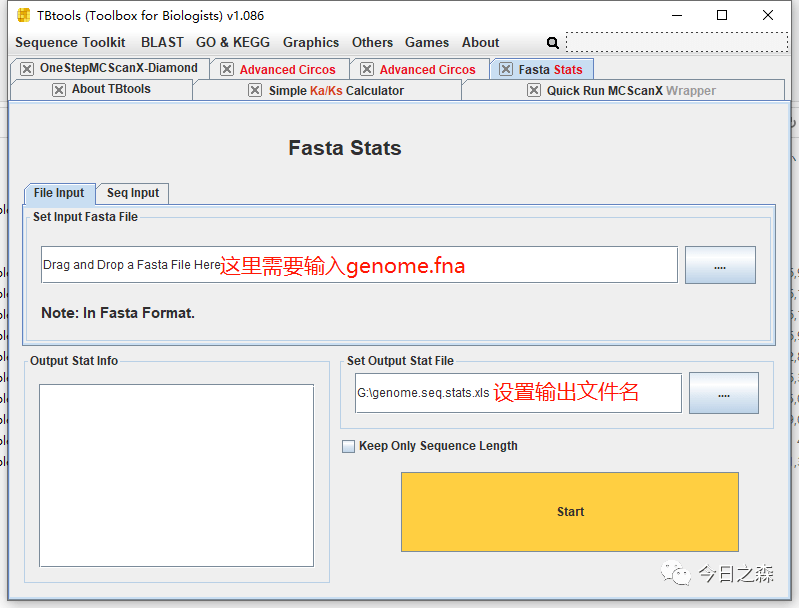
生成的genome.seq.stats.xls打开后将Simplified_ID和Length两列信息保存为ChrLen.txt。
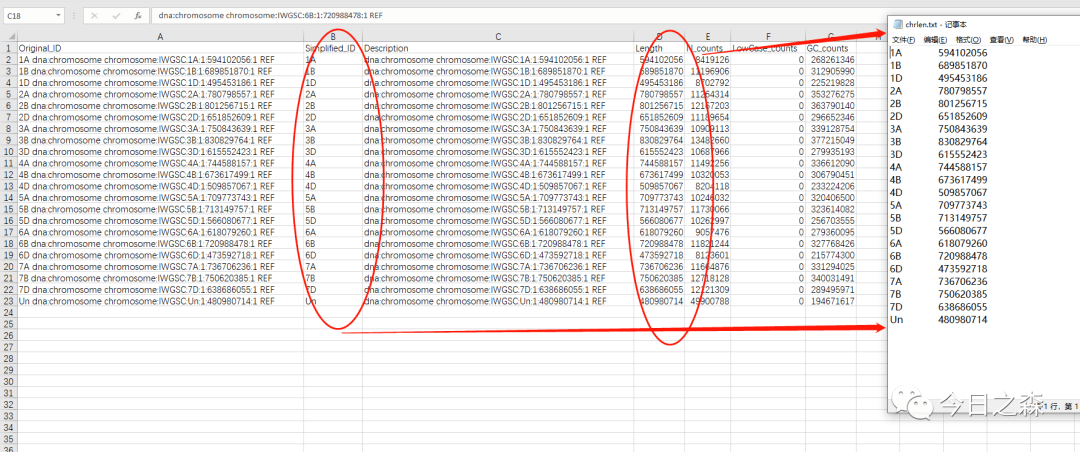
到此为止,就准备好了第一个文件ChrLen.txt。
第二步:先来准备第3个文件(基因间的关联文件)
这部分功能超级强大,重点在快,超快!快到什么程度呢,小麦这种如此庞大的基因组自身比对到自身只需要40min!
打开如下界面,并按要求设置输入文件和输出路径。
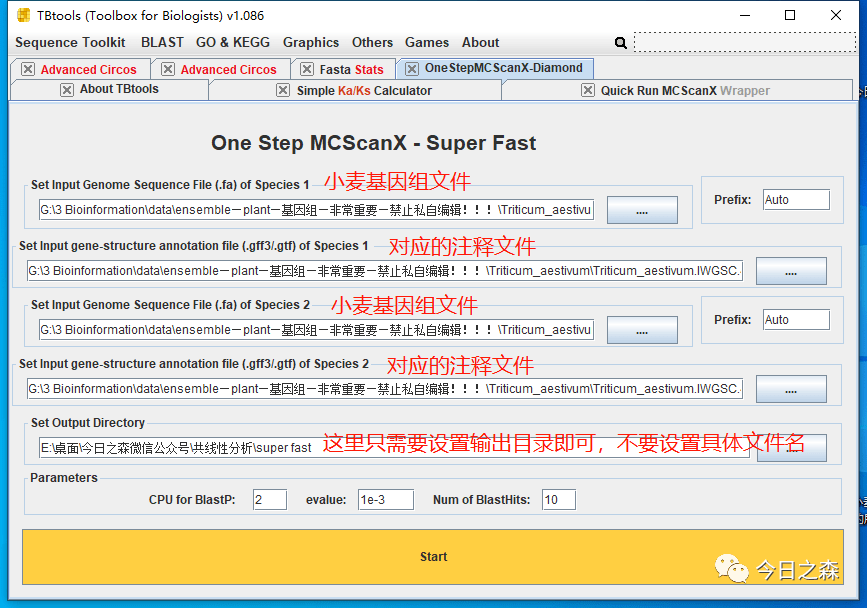
小麦基因组自身比对到自身只需要40min!!!(其他小基因组物种间的比对可能分分钟就搞定了)
start之后输出以下文件。
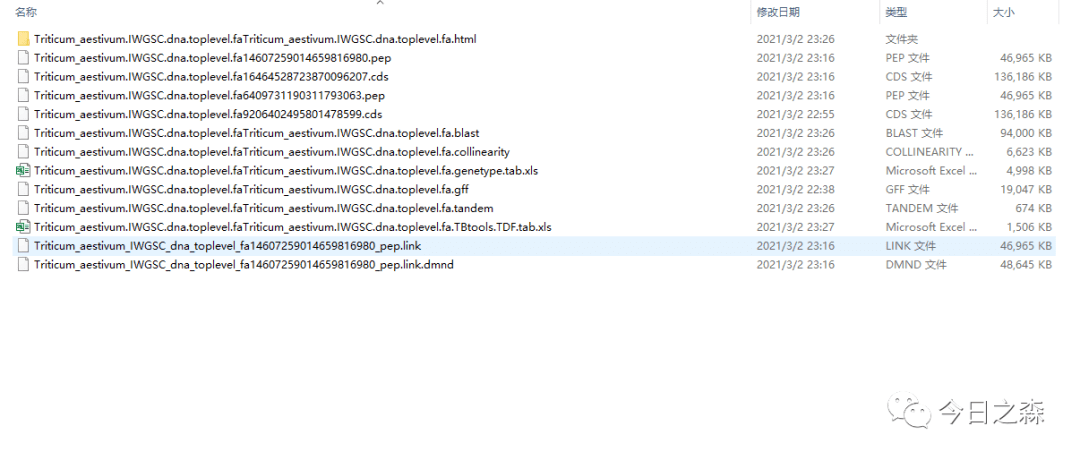
MCScanX运行结束后已经解决了大部分问题,后面就很简单了。
首先从上述结果中的.collinearity生成GenePair.tab.txt。
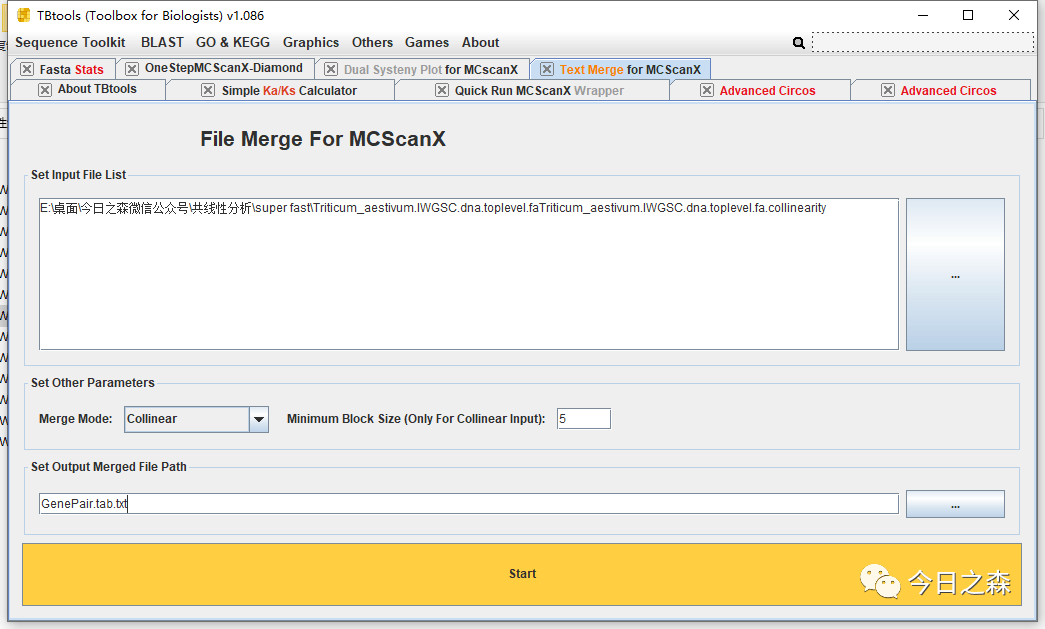
然后用 GenePair.tab.txt生成linkedRegion.tab.txt。
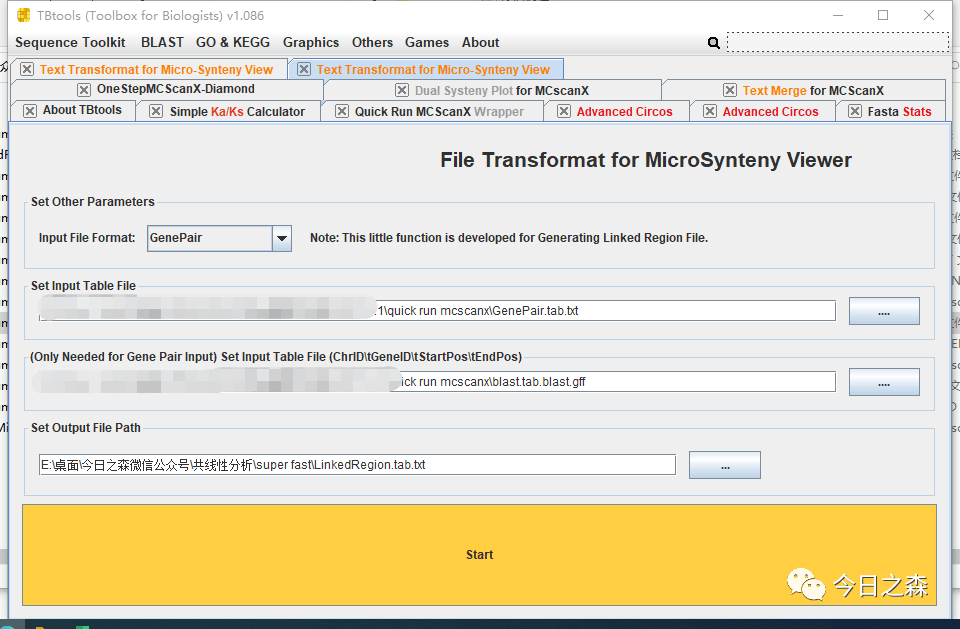
输入文件准备齐全,就可以绘图了。
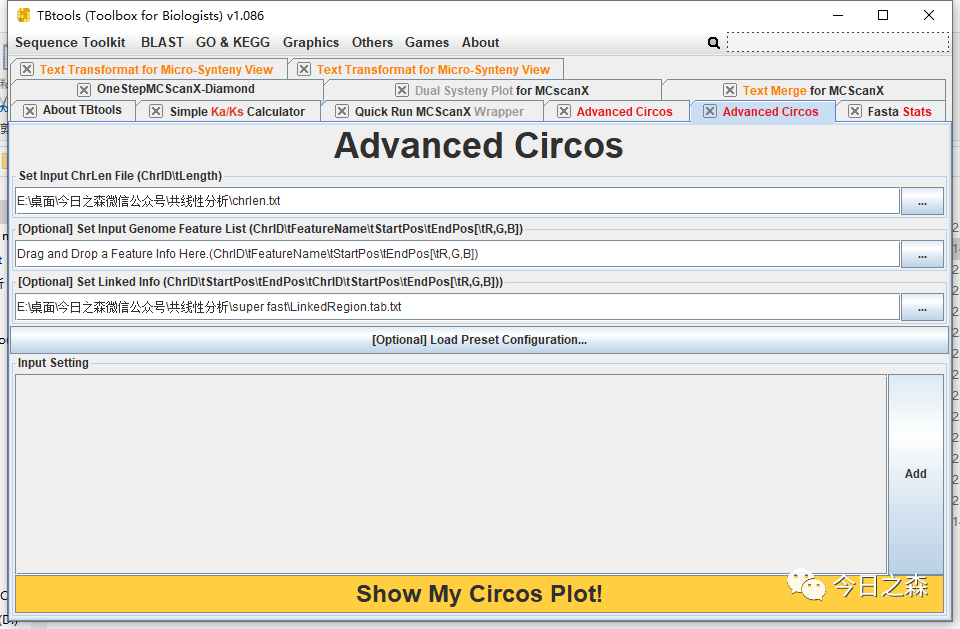

稍微调整一下。
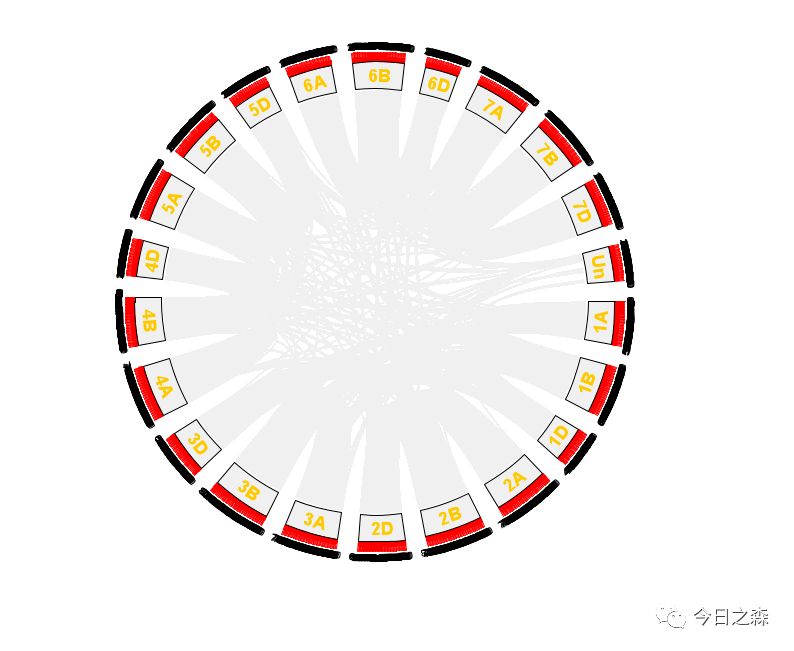
到这里很明显还没有结束,因为很多时候需要在上图中展示某些gene family内部参与的复制事件,因此,我们需要在上图将这些基因相关的连线高亮显示。
第三步:准备第2个文件
首先需要从上文得到的串联基因对中提取gene family成员相关基因对。
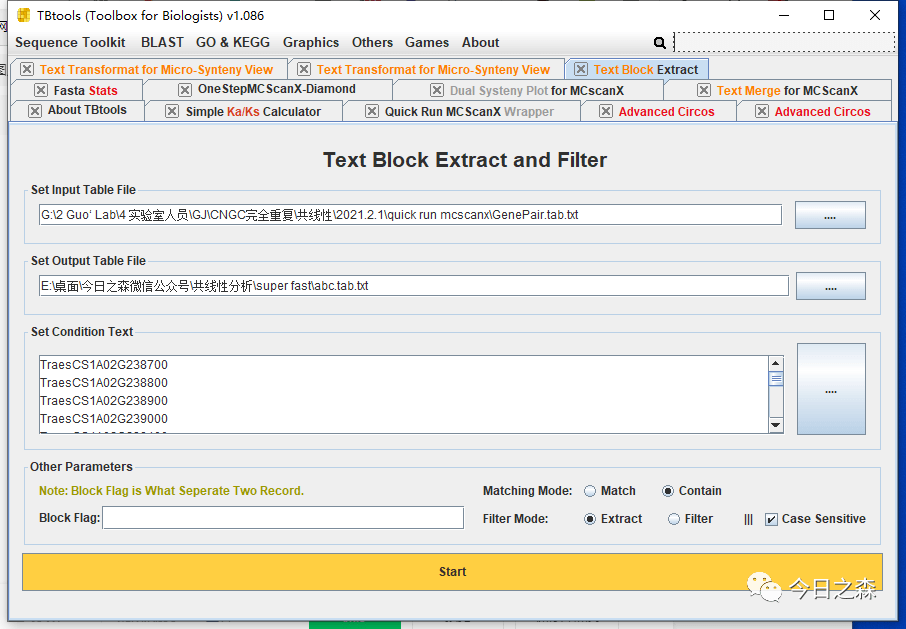
然后根据前述操作,获得基因家族内部的LinkedRegion文件。
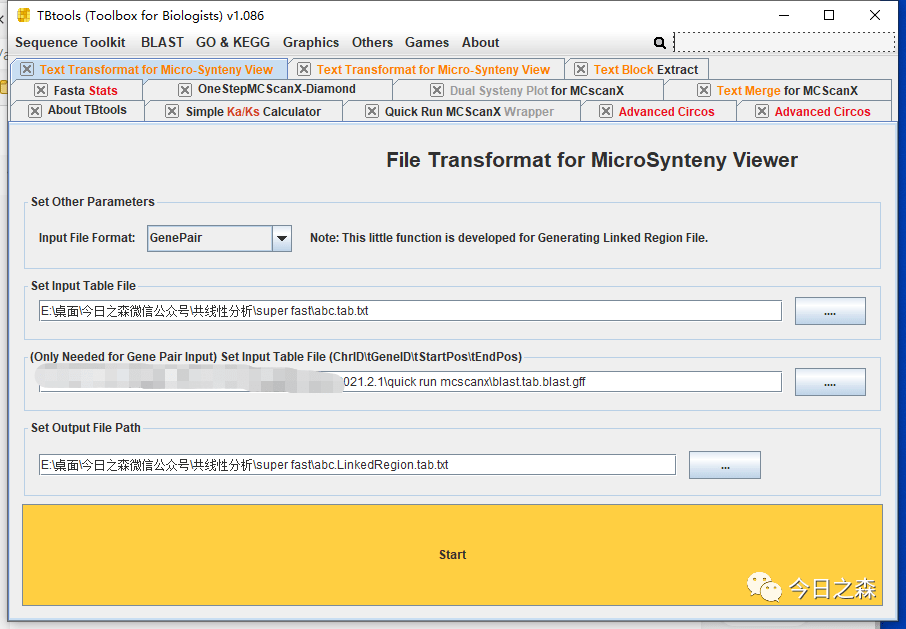
然后。打开abc.LinkedRegion.tab.txt,在最右边一列加入RGB颜色。
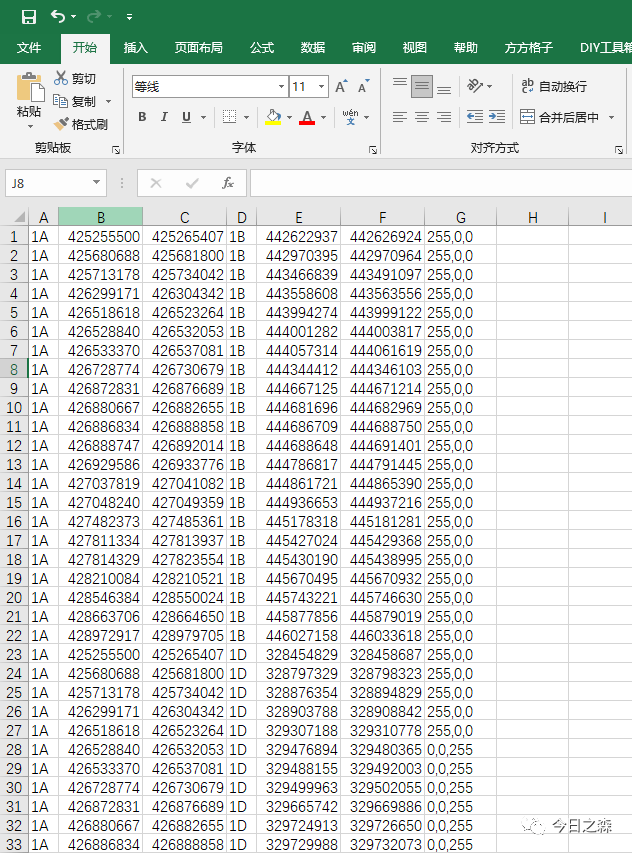
然后,将这个abc.LinkedRegion.tab.txt与基因组linkedRegion.tab.txt合并后重新绘图。如下。(由于这里是从开头随意取的基因,并非基因家族。因此图可能奇怪)
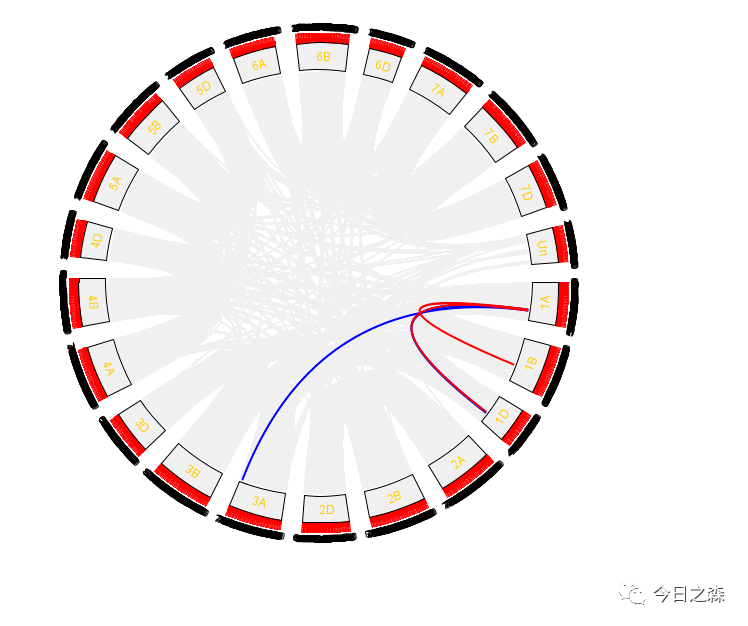
接下来,还需要gene ID显示。
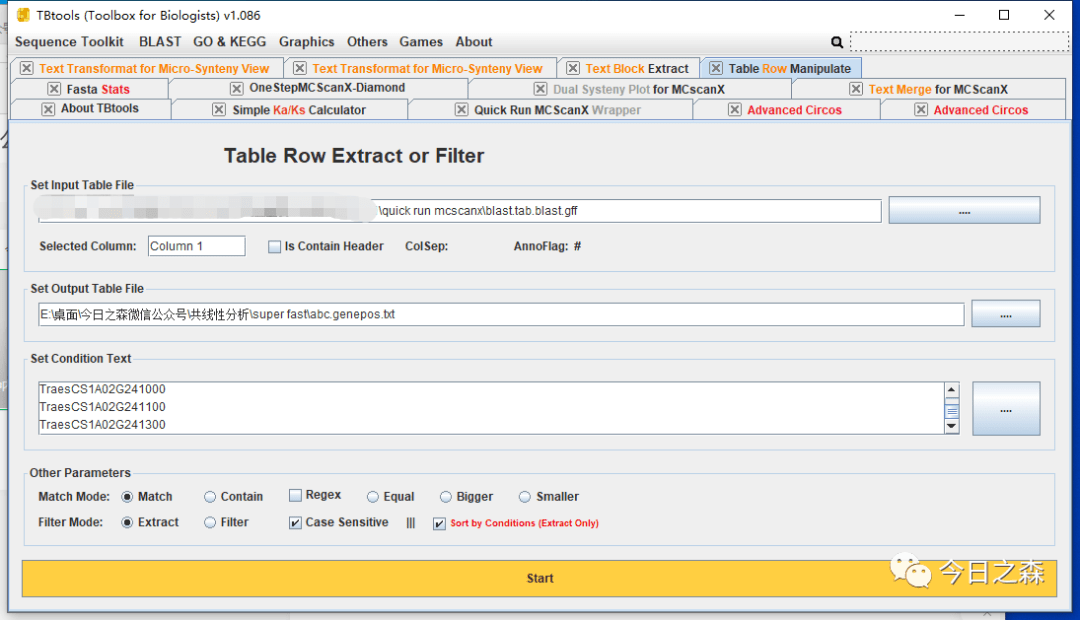
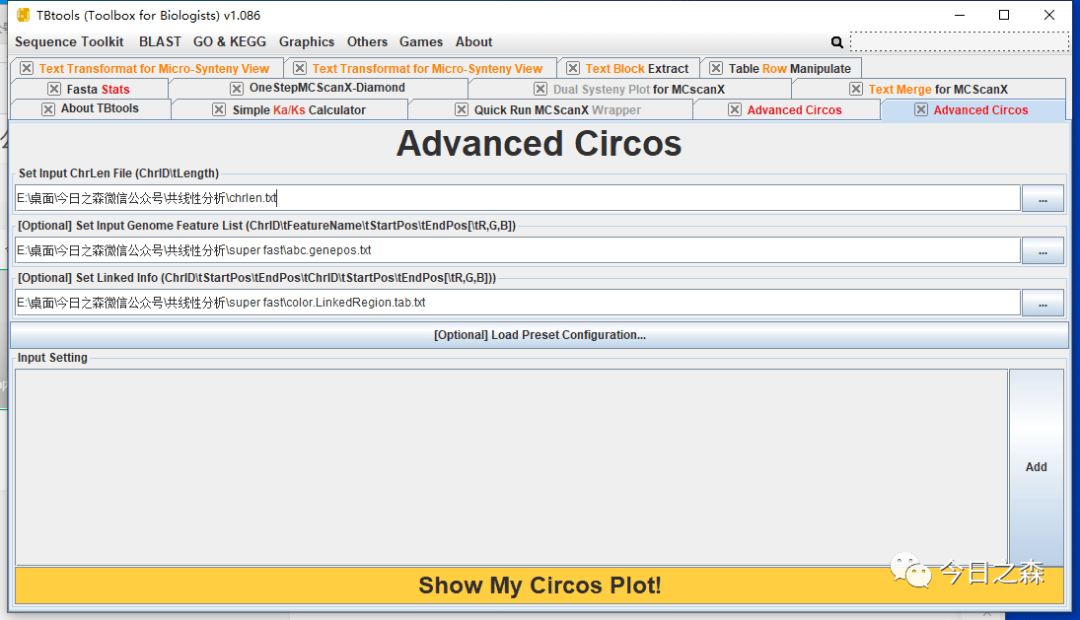
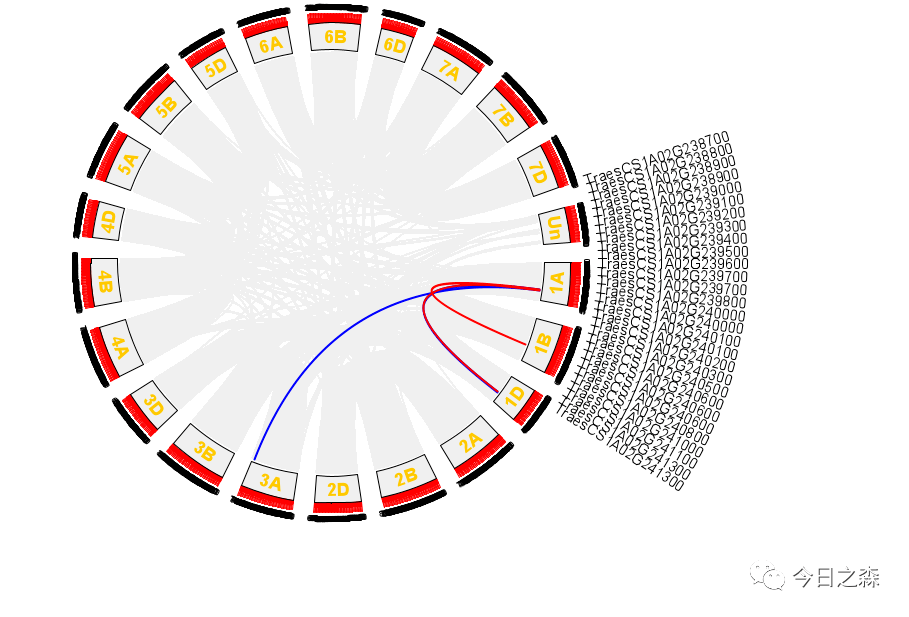
对于物种内共线性circus的绘制,大致如此。
5.3.2 物种间共线性分析
早先写过物种内共线性circus——作图步骤 ,但是有不少朋友还是在后台留言,无法完全重复。我仔细看了之后发现,的确是我在有些细节上写的不够详细。但是不想去完善了。毕竟tbtools的实现逻辑已经很简单了。
那就是IOS,即input,output,start。
最近又有读者在做物种间共线性,其实和物种内也没差。那今天就写一下物种间的共线性circos作图。
这篇推文不只是演示物种间的共线性,而是更进一步让大家体会使用tbtools时需要注意的问题。以便指导大家下次使用tbtools其他功能时能够具备自行解决和筛查可能出现的报错原因。
看到下图即可知道需要准备哪些文件。(这一步不要急着操作,往下看,看完再说)
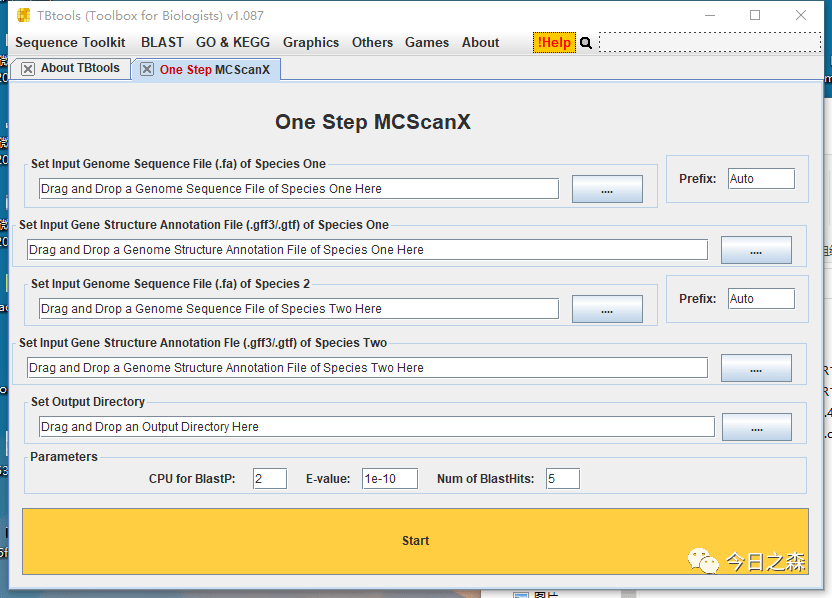
第一步:准备物种1和物种2基因组序列文件和注释文件。
然后打开One Step MCScanX(这一步不要急着操作,往下看,看完再说)
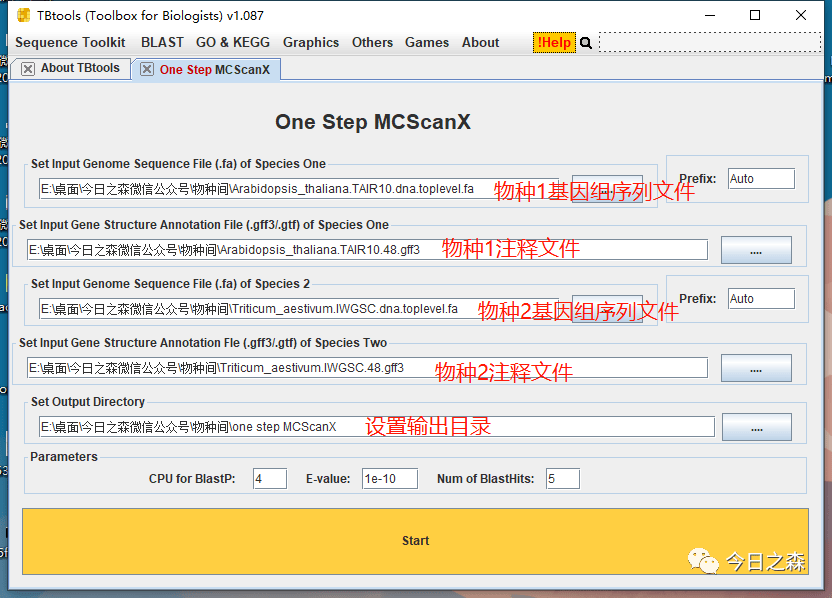
start之后,通常会有黑框弹出。不用管,继续等待。
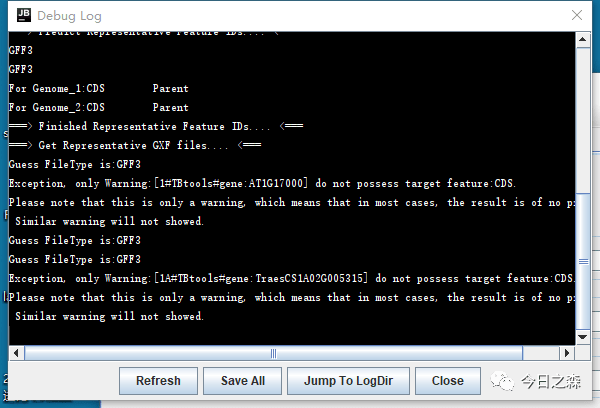
11:37开始运行。
中午电脑在运行,我就去吃饭了。晚上七点多过来一看卡死了。突然想起来早上的上一步出了点小状况。
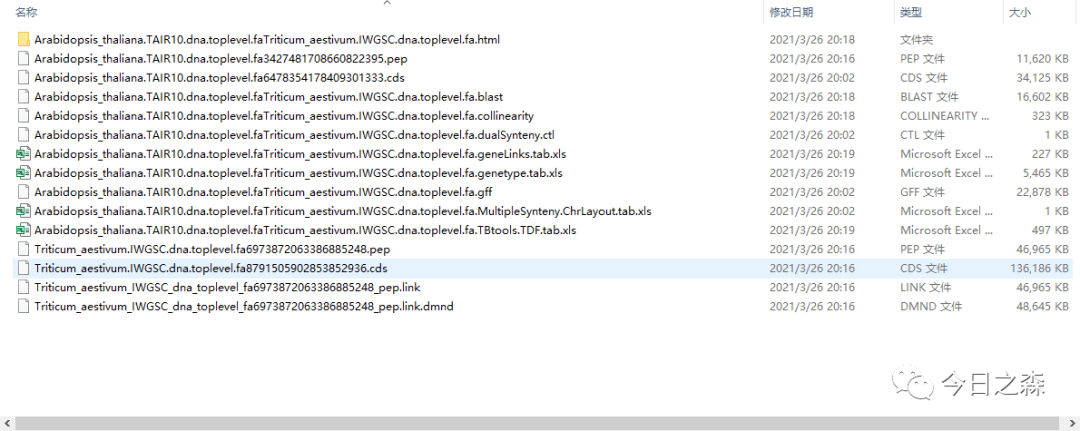
好,及时纠正。突然想起来有个super fast的插件。打开 One Step MCScanX-Super Fast,真的超快, super fast!!!

大约等待20min就finished。输出文件如下:
下面出图
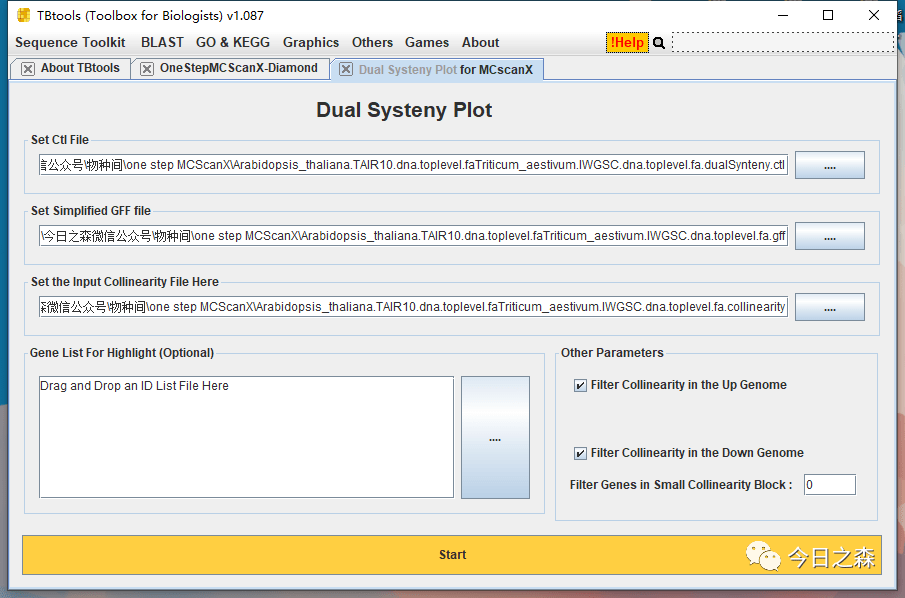
然后,同样需要高亮一些我们需要展示的基因,我这里就从头取20个基因,用以展示。
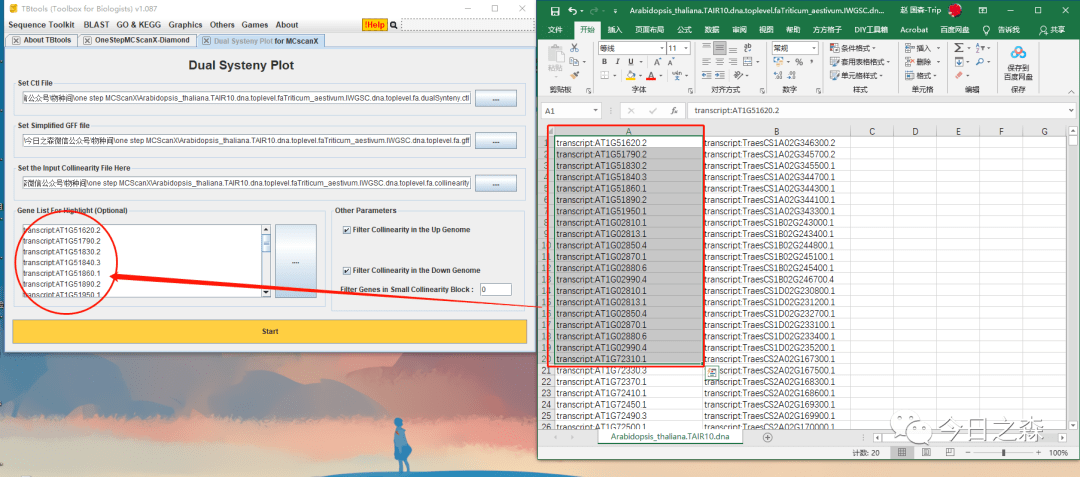
到这里,两物种间的共线性就做好了。
下面我们接着看多物种间的共线性怎么做?
上面对拟南芥和小麦进行了共线性。由于我电脑上之前下载过马铃薯的,而且马铃薯基因组相对小很多。操作起来会快一点,所以我们就对马铃薯和拟南芥进行共线性。
同样。
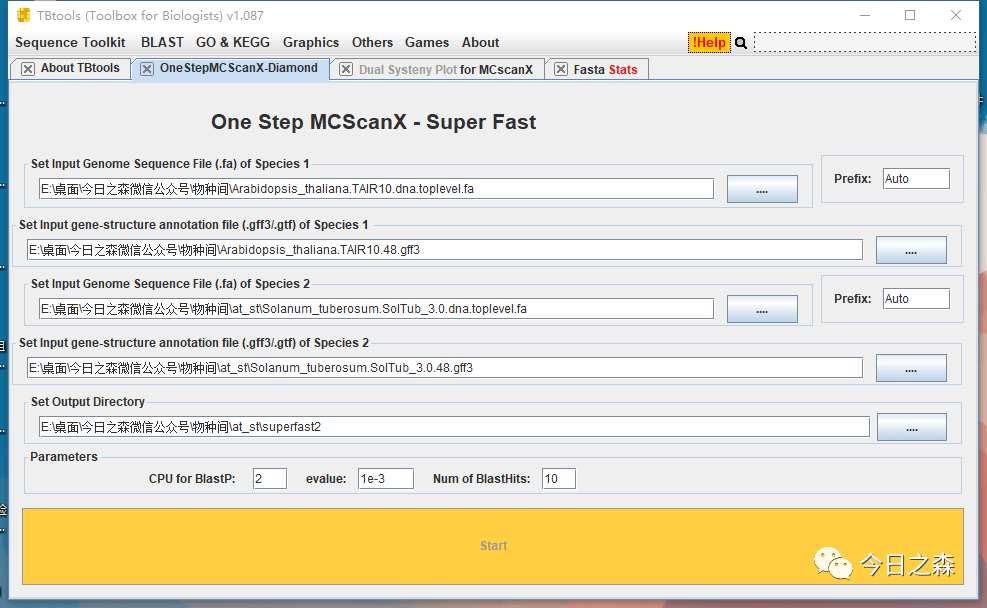
如果弹出黑框,请忽略,基本不影响。
21:22start
21:24finished
输出文件如下:
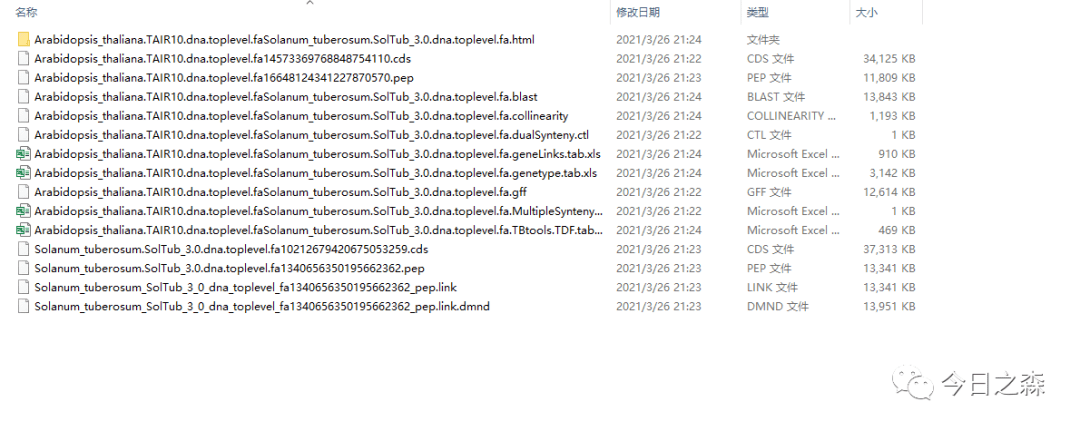
下面我们来作图
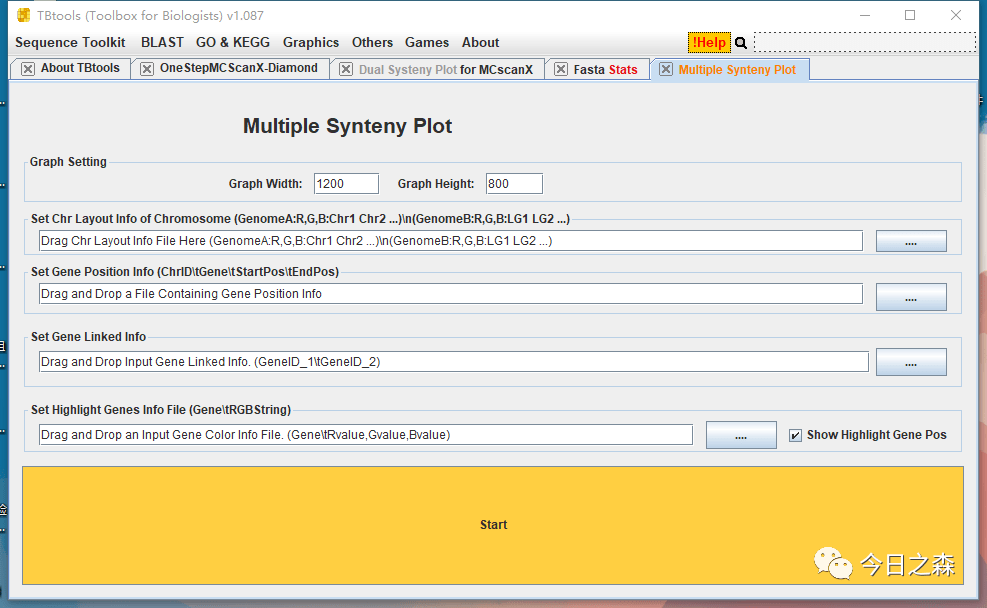
看到这个界面,就知道我们需要准备哪几个文件。
好,那就合并这几个文件。然后出图。
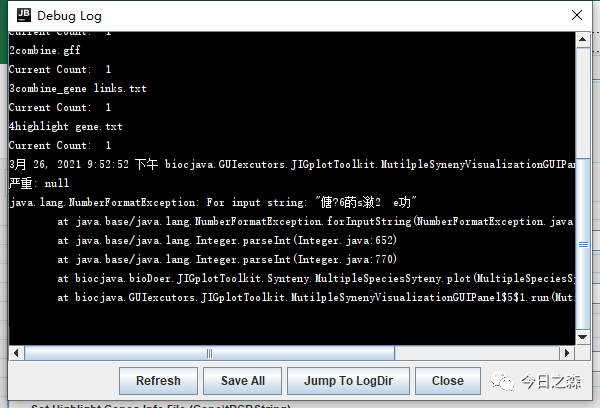
显然,肯定会报错。刚才两两比对的输出文件肯定没有错。那问题出在哪里呢?由于我们在操作之前没有对3个物种的染色体ID进行重命名。由于拟南芥和马铃薯的染色体都是以数字1、2、3……进行命名的,因此在后续分析和作图过程中肯定会报错。
----------------------------强制分割线-------------------------------------
【下面正式进入多物种间共线性】
第一步:数据准备
好,那就重新来过,对染色体ID进行统一。

然后准备出图,这里需要对两次各自比对的文件进行合并整理。具体看个人了,这里不进行演示。有需要的后台私信。
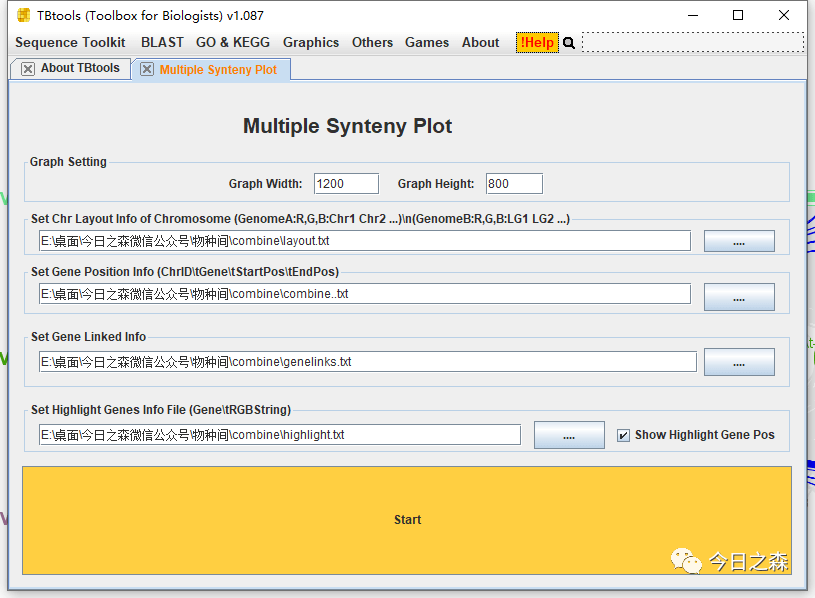
出图如下:(这里只选取了前拟南芥前100个基因进行高亮显示)
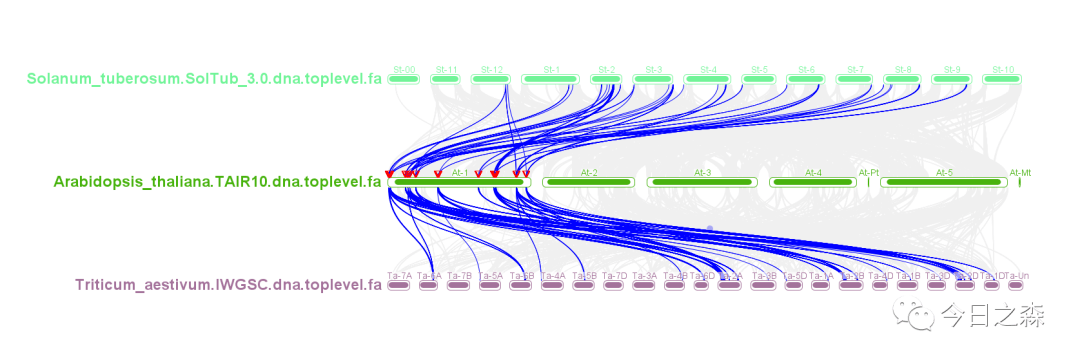
大致就是如此。但仔细一看,除了拟南芥染色体是按顺序排列的,St和Ta的染色体是乱序的。这就需要简单调整一下了。
这样看起来就好多了。
当然对于这个图还有很多地方可以调整。
5.4 GO富集分析
……
5.5 KEGG分析
https://www.kegg.jp/kegg/pathway.html
软件安装多重序列比对软件mafft的安装及使用
构建进化树过程中,我们常用到MEGA软件集成中的Muscle等进行多序列比对,这针对小数据集(几百条序列以内)的进化树构建或许还是可以的,针对大数据集(几千条序列以上)的进化树构建就不现实了。
由于大多数集成软件在面对大文件时,或多或少会面临运行卡顿,甚至使电脑卡死,或者根本无法运行大文件。
比如Windows自带的记事本根本打不开几十Mb以上的文本文件,我们常用的基因组蛋白数据大都在几Mb至几十Mb,根本无法操作,小麦基因组数据甚至有十几个G那么大。因此针对这些大文本文件再次推荐使用Notepad软件进行操作,毕竟大多数文件我们要能打开并看到具体内容才放心。
MEGA面临的问题也是一样的,我们在进行几百甚至上千条序列比对时,速度之慢对于大多数人来说实在等不及,慢就慢吧,由于实在太慢,很多时候在中途就会导致电脑死机等突发情况,再加上很多时候几千条需要至少需要2天以上的时间,如果遇到突发停电等情况那就无疑是给满心期待,等待比对结果的同学浇了一盆凉水,因为要重新来过。
这时候,就需要多序列比对利器mafft软件了。
mafft软件官网 https://mafft.cbrc.jp/alignment/software/windows.html
(网站今天好像打不开,后台回复 mafft 即可快速获取)
下面对mafft软件的使用进行说明并操作
mafft软件不需要安装,进入官网下载Windows版本的软件压缩包,解压后双击mafft.bat文件即可使用。
双击后界面如下:
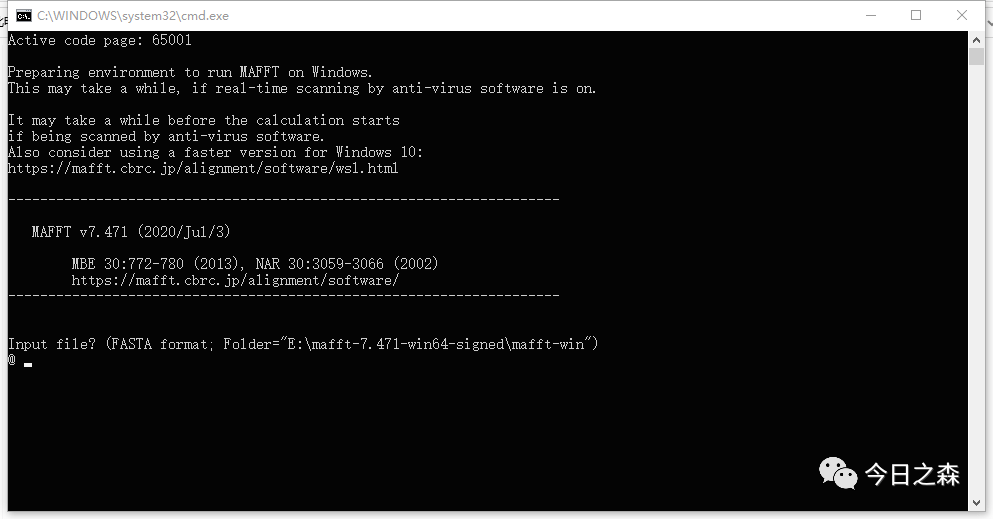
这里需要注意一点:如果下载后想直接在解压目录下使用,则需要将fasta格式的蛋白序列文件也放在该目录下,如下面这样:
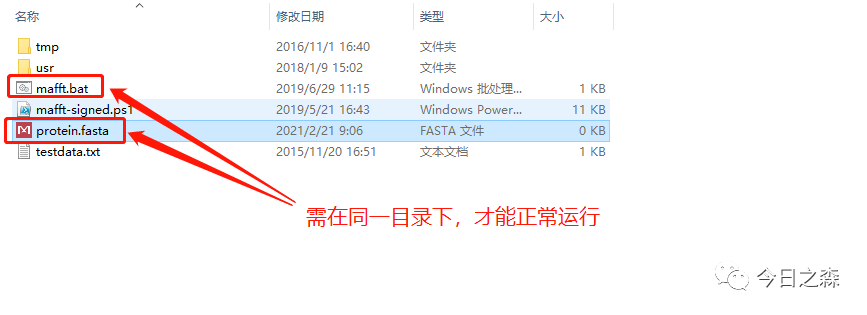
但是每次都把待比对的fasta格式的蛋白序列文件往这里放也很麻烦。聪明的你肯定在想,如果能在任何目录下都能随时调用mafft软件就好了。这就需要对mafft软件进行环境配置,配置成功后就可以在任何目录下都能轻松调用mafft软件。
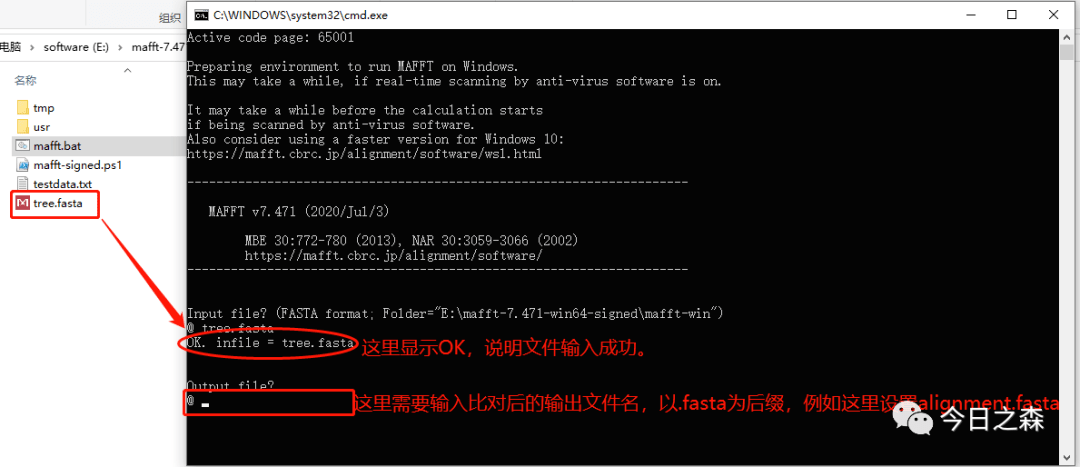
- Input file? 需要输入待比对的文件名,这里使用之前的hsp20数据进行操作,输入文件名后按Enter进入下一步。如下图:
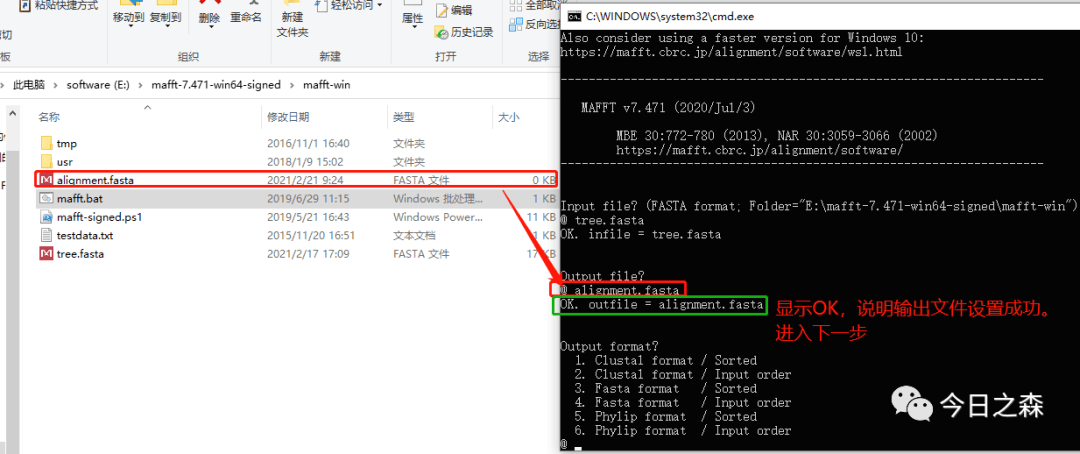
2.output file?
需要输入比对后的输出文件名,以.fasta为后缀,例如这里设置为alignment.fasta。(注:这个输出文件需要提前新建并命名为alignment.fasta后才可以输入)。如下图:
3.output format?
输出格式,这里选择fasta格式,所以输入3,然后Enter。如下图
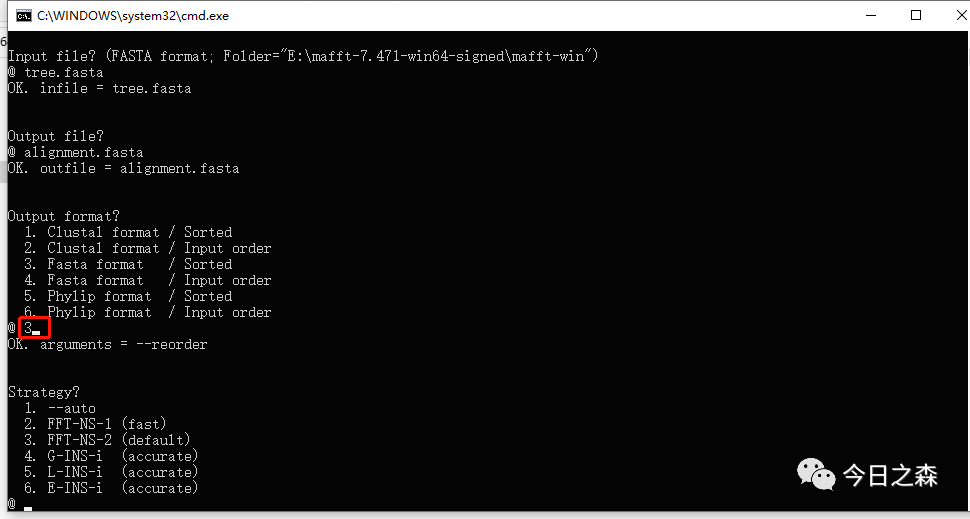
4.Strategy?
算法,选择1会自动选择最优算法,其他的算法在括号中有简单介绍,具体的区别可以查看官网的说明文件。这里我们输入1,然后Enter。如下图:
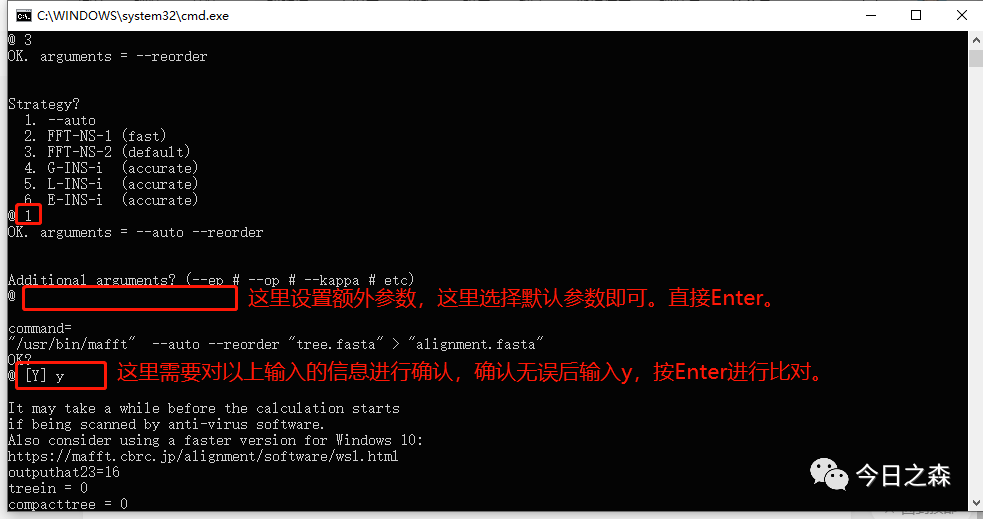
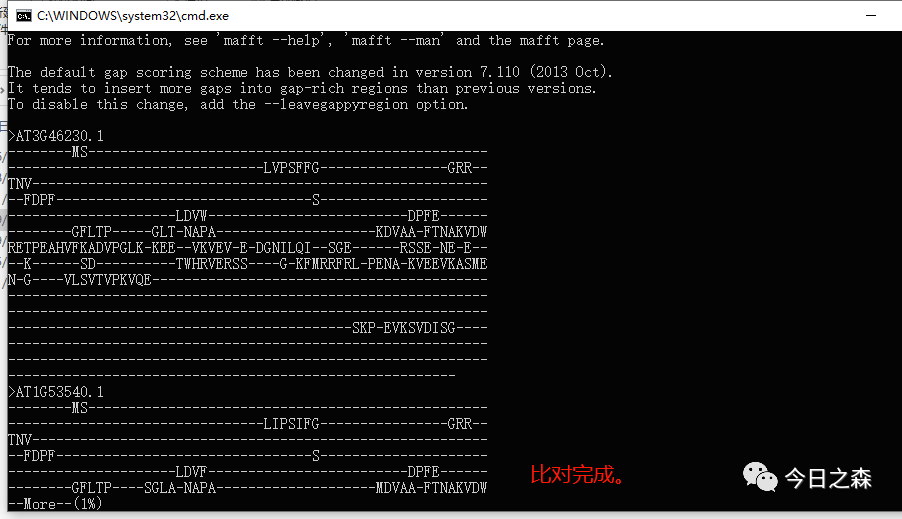
5.稍等几秒,出现如下界面则说明比对完成。如下图:
我们这里使用了约80条序列,比对完成用时约5s。
6.可以看到,刚才新建的空白alignment.fasta已经有了内容。如下图:
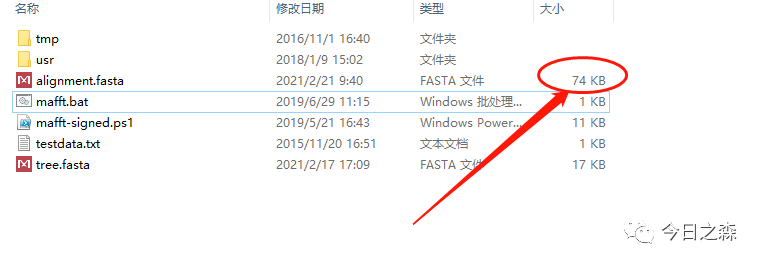
7. 接下来就可以用这个比对好的文件alignment.fasta进行建树了。
可以使用MEGA,Fasttree等建树软件进行建树了。
当然,如有需要,可以进一步对比对结果进行修剪。
到这里,相信对于mafft的使用方法有了大致了解,详细信息可以进入官网查看。
问题来了:mafft会用了,可是mafft比对结果的准确度到底如何?
可以参考下面两篇文献深入了解。
最后,如果满足这个条件
Important note:This tool can align up to 500 sequences or a maximum file size of 1 MB.
可以使用这个在线mafft
适用于大数据集进化树构建的Fasttree安装及使用
进化树构建的软件很多,但同样面临运行大数据集的文件时会出现软件未响应情况。另外很多时候我们需要针对同一序列文件不止一次建树,如果每次都耗时过长,无疑会大大降低我们的工作效率。
我之前对约2000条序列使用MEGA构建进化树,耗时约24h。这个时间虽然很快了,但还是有点等不起。
于是昨晚尝试使用Fasttree对约7000条序列构建进化树,耗时约4h30min。这就很快了。
下面还是使用hsp20基因的alignment.fasta进行操作。
Fasttree软件官网
http://www.microbesonline.org/fasttree/
下面开始操作:
Fasttree不需要安装,下载后即可使用
- 鼠标停在fasttree.exe目录中,按下WIN+R键 输入cmd,打开cmd窗口。
- 下面需要将工作目录定位到当前文件夹下,我这里是E:\桌面\今日之森微信公众号\基因家族2\fasttree 所以先输入E: 按Enter进入E盘。
- 然后输入cd E:\桌面\今日之森微信公众号\基因家族2\fasttree ,按Enter,进入当前工作目录。
- 下面就可以使用Fasttree建树了。 下面是fastatree的使用说明:
FastTree < alignment_file > tree_file orFastTree alignment.file > tree_file Usethe -wag or-lg options tousethe WAG+CAT orLG+CAT modelinstead. You can also useyour own transition matrix
Toinfer a tree fora nucleotide alignment withthe GTR+CAT model, use
FastTree -gtr -nt < alignment.file > tree_file orFastTree -gtr -nt alignment_file > tree_file Ifyou donotspecify -gtr, thenFastTree will usethe Jukes-Cantor + CAT modelinstead. Usethe -gamma option(about 5% slower) ifyou want torescale the branch lengths andcomputea Gamma20-based likelihood. Gamma likelihoods aremore comparable across runs. These also allowforstatistical comparisons ofthe likelihood ofdifferent topologies ifyou usethe - loglogfileoption(see details). The changeinthe scale ofthe tree isusually modest ( 10% orless).
Ifyou areusingWindows, run FastTree withinthe command-line environment ( useStart/ Run / "cmd"). Ifyou areusingMacOS orLinux, you may need tospecify the pathofthe executable byusing"./FastTree"rather than"FastTree".
Tosee what versionofFastTree you have andforinformation onFastTree 's options, run FastTree without any arguments or with the -help option.
安装上述使用说明,那我们这里就运行以下代码进行建树,其中hasp20.nwk为输出文件名。
运行结束,耗时约5s。
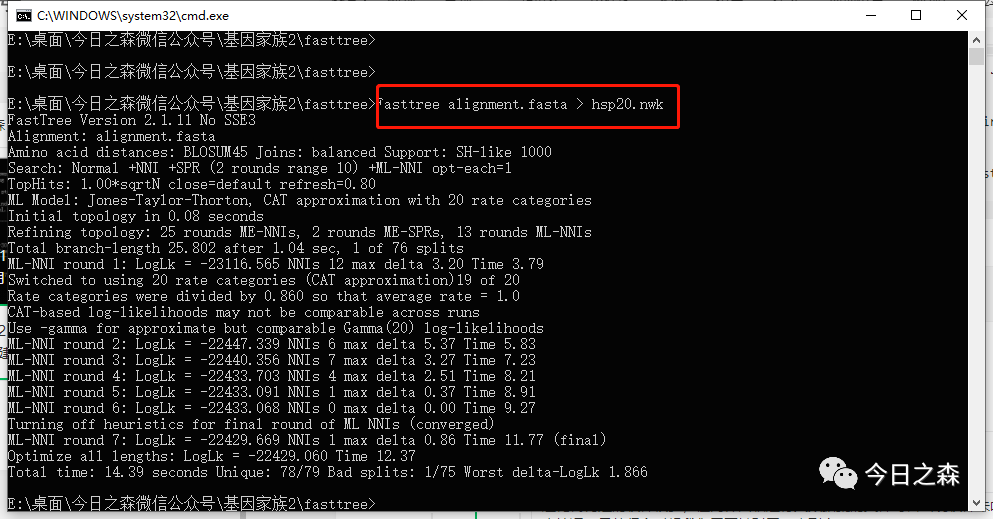
打开文件夹,可以看到hsp20.nwk树文件已经生成。
如果需要使用其他算法,则按上述说明文件运行相应代码即可。
转自:今日之森- 本文固定链接: https://maimengkong.com/kyjc/937.html
- 转载请注明: : 萌小白 2022年5月26日 于 卖萌控的博客 发表
- 百度已收录
