如果大家想要了解测序原理,参考文章( 测序产生了那么多数据,你知道测序的原理吗? ); 如果大家想要了解测序数据格式,参考文章()
本文介绍RNA-seq的具体分析流程。
1、cutadapt去接头
我们拿到的测序数据一般是带有接头的fastq格式文件,需要用cutadapt把接头去掉。具体代码如下:
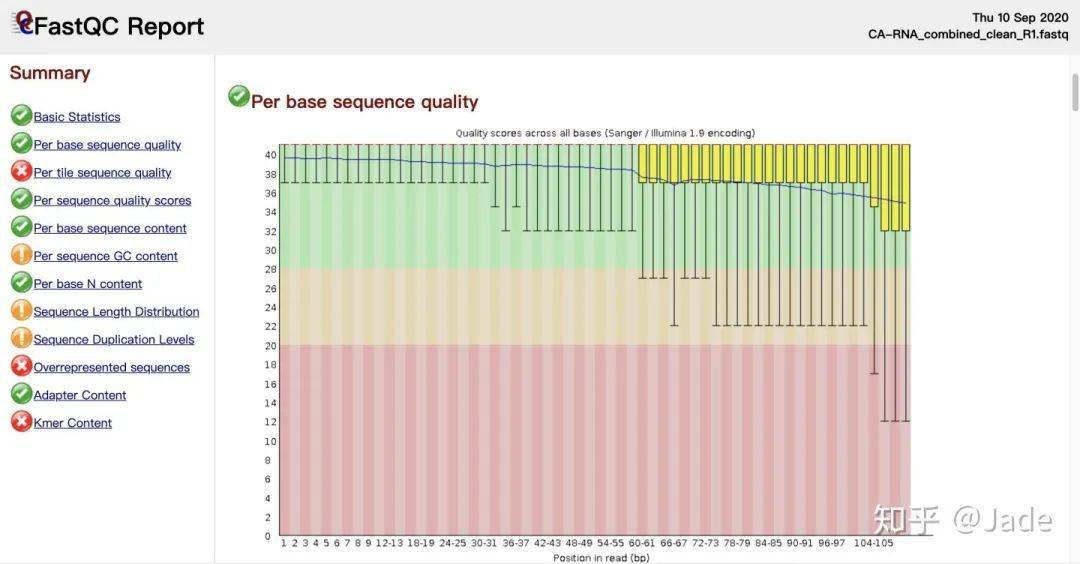
一般做完去接头会用fastqc查看数据质量是否满足要求。
fastqc结果如下所示:
2、bowtie2建索引
我们需要对下载的参考基因组建立索引,代码如下:
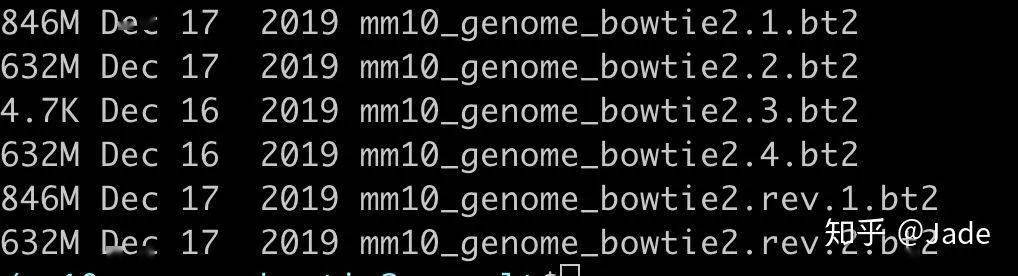
mm10的bowtie2结果,第一列为文件大小 3、tophat2比对
把我们的测序序列比对到参考基因组上。代码如下:
4、cufflinks定量
对比对后到bam文件定量,即确定每个基因的表达值。代码如下:
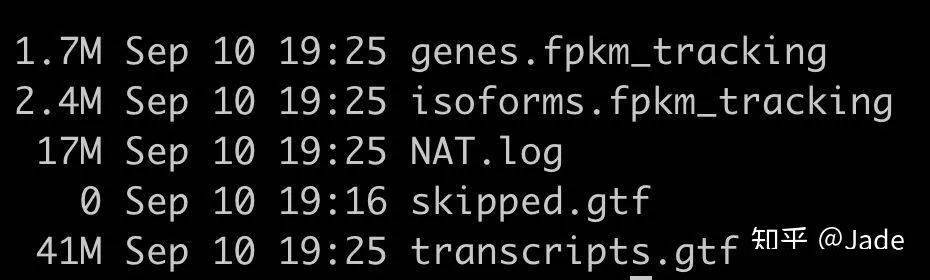
1、cufflinks组装结果

2、cuffmerge合并结果
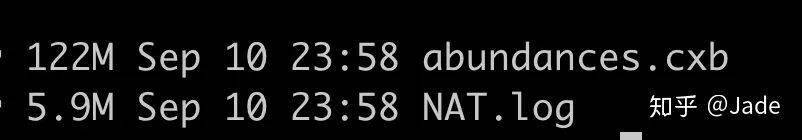
3、cuffquant定量结果
4、cuffdiff差异分析结果
5.接下来用r做差异表达基因
##Tools preparation if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install("DESeq2") library("DESeq2") ##The count data preparation mode:character raw_data<-read.table(".../fpkm.txt", header = TRUE) ##convert data.frame to matrix raw_data_mat<-as.matrix(raw_data) ##convert "character" matrix to "numeric" matrix raw_data_mat2<-apply(raw_data_mat, 2, as.numeric) ##delete the first column which is extra raw_data_mat2<-raw_data_mat2[,-1] ##add rownames in the gene name of "raw_data_mat" rownames(raw_data_mat2)<-raw_data_mat[,1] ##check matrix mode is.numeric(raw_data_mat2) ##save "raw_data_mat2" as "count_data" count_data<-raw_data_mat2 ##the number should be integer which the next DESeq2 requires count_data_round<-round(count_data) ##check whether exist duplicated row or it will go wrong anyDuplicated(row.names(count_data) ) ##delete duplicated row in count_data count_data_dedup<-count_data[!duplicated(row.names(count_data)), ] ##check whether exist duplicated row in count_data_dedup anyDuplicated(row.names(count_data_dedup) ) ##make a dds(three replicate) condition<- factor(c("ctrl","ctrl","ctrl", "test","test","test") ) coldata<- data.frame(row.names = colnames(count_data[,1:6]), condition) dds <-DESeqDataSetFromMatrix(round(count_data_dedup), coldata, design= ~ condition) ##DESeq dds2<-DESeq(dds) #resultsNames(dds2) res<-results(dds2) ##pick differential genes or use " diff_gene_deseq2 <-subset(res, padj < 0.05 & abs(log2FoldChange) > 1) " diff_gene_deseq2<-subset(res,padj < 0.05 & (log2FoldChange > 1 | log2FoldChange < -1)) ##merge differential gene data and normalized raw_count data result_data<-merge(as.data.frame(diff_gene_deseq2), as.data.frame(counts(dds2, normalized=TRUE)), by="row.names", sort=TRUE) ##save result file write.csv(result_data,file = "..../ctrl_test")如果大家不仅想要差异表达基因,还想知道样本的SNP和INDEL,可参考这篇文章()
拿到SNP和INDEL后如果做注释,可参考这篇文章
- 本文固定链接: https://maimengkong.com/kyjc/723.html
- 转载请注明: : 萌小白 2021年7月26日 于 卖萌控的博客 发表
- 百度已收录
