今天要介绍一下数据挖掘,从大量已有的数据来产生新的hypothesis。正如我在ChIPseeker的文章里写的:
There are increasing evidences shown that combinations of TFs are important for regulating gene expression (Perez-Pinera et al., 2013; Zhu et al., 2008). However, systematically identification of TF interactions by ChIP-seq is still not available. Even if a specific TF binding is essential for a particular regulation was known, we do not have prior knowledge of all its co-factors. There are no systematic strategies available to identified un-known co-factors by ChIP- seq.
并没有方法可以大规模地预测未知的共同调控因子,而数据挖掘就是要给我们这种预测的能力。
我当年在写ChIPseeker的时候,我有纠结是写篇Bioinformatics的application note呢,还是写篇长文灌水NAR,毕竟NAR影响因子高一点,最后还是发了Bioinformatics,因为我没钱,囧,Bioinformatics不要版面费啊。然后限于篇幅,ChIPseeker有大量可视化的函数,我在文章中一张图都没放!!!如果当时决定发NAR的话,这个数据挖掘这一块我就会写多点。
做注释在Windows上有个软件CisGenome,是Hongkai Ji课题组做的,他们还做了一个hmChIP的database:
这个hmChIP可以说是先行者,它只支持人和鼠,收集了一些GEO的数据,这个数据库做得比较早,当年GEO上的ChIPseq数据并不多,当由于搞得早,也发了一篇Bioinformatics,当然灌水之后,就不维护了,数据没更新,而基因组版本号还停留在hg18和mm8之上,所以即使它还能用,我想也不会有人去用了。这个数据库做一个事情,你可以上传自己的数据,然后它会和数据库中的数据进行比较,给你报道出你的数据和数据库中的数据overlap了多少,仅此而已。
可以说这是一个雏形,我一直在想overlap了多少,不是很能说明问题,多少算多?能不能算个p值出来,overlap了这么多,随机情况下是不太可能产生的。思来想去,没办法直接算啊,于是求助于permutation,没有枪没有炮,我们自己造。
写了一个shuffle函数,也就是传说中的洗牌,你给基因位置信息,随机给你返回等长的片段,但位置改变了。
p <- GRanges(seqnames=c("chr1", "chr3"),
ranges=IRanges(start=c(1, 100), end=c(50, 130)))
shuffle(p, TxDb=txdb)
## GRanges object with 2 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 [239651460, 239651509] *
## [2] chr3 [163562934, 163562964] *
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths 有了这个函数,我们可以对数据洗牌洗它一千次、一万次,那么就可以计算一千一万个overlap,来形成背景分布,实际数据的overlap在这一背景分布中产生的概率就可以计算出来。
enrichPeakOverlap(queryPeak = files[[5]],
targetPeak = unlist(files[1:4]),
TxDb = txdb,
pAdjustMethod = "BH",
nShuffle = 50,
chainFile = NULL,
verbose = FALSE)
## qSample
## ARmo_0M GSM1295077_CBX7_BF_ChipSeq_mergedReps_peaks.bed.gz
## ARmo_1nM GSM1295077_CBX7_BF_ChipSeq_mergedReps_peaks.bed.gz
## ARmo_100nM GSM1295077_CBX7_BF_ChipSeq_mergedReps_peaks.bed.gz
## CBX6_BF GSM1295077_CBX7_BF_ChipSeq_mergedReps_peaks.bed.gz
## tSample qLen tLen N_OL
## ARmo_0M GSM1174480_ARmo_0M_peaks.bed.gz 1663 812 0
## ARmo_1nM GSM1174481_ARmo_1nM_peaks.bed.gz 1663 2296 8
## ARmo_100nM GSM1174482_ARmo_100nM_peaks.bed.gz 1663 1359 3
## CBX6_BF GSM1295076_CBX6_BF_ChipSeq_mergedReps_peaks.bed.gz 1663 1331 968
## pvalue p.adjust
## ARmo_0M 0.90196078 0.9019608
## ARmo_1nM 0.25490196 0.4444444
## ARmo_100nM 0.33333333 0.4444444
## CBX6_BF 0.05882353 0.2352941
函数里有一个chainFile的参数,如果你提供了,数据的比较可以跨基因组版本,甚至于可以跨物种比较。
有了这个函数,那么我再打通GEO,让用户可以检索下载GEO的数据,如此一来,你就可以用自己的数据去和GEO的数据做比较,或者就可能挖出overlap非常significant但没被报道到的co-factor呢!
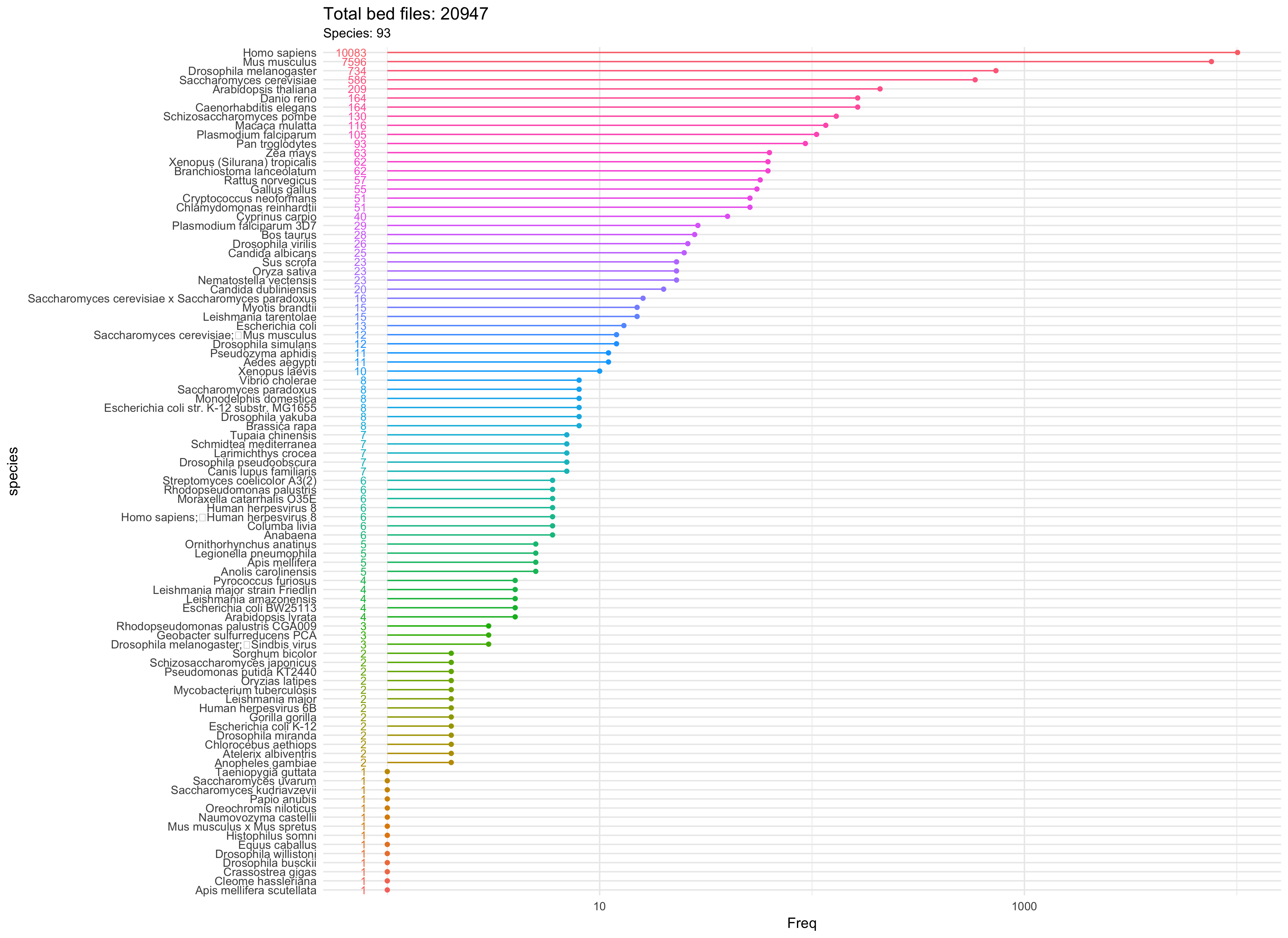
ChIPseeker已经收集了超过90个物种,超过2万个BED文件的信息。你可以用getGEOgenomeVersion列出所有支持的基因组及相应的BED文件数目。
getGEOgenomeVersion()
## organism genomeVersion Freq
## 1 Anolis carolinensis anoCar2 5
## 2 Bos taurus bosTau4 2
## 3 Bos taurus bosTau6 24
## 4 Bos taurus bosTau7 2
## 5 Caenorhabditis elegans ce10 4
## 6 Caenorhabditis elegans ce6 64
## 7 Danio rerio danRer6 6
## 8 Danio rerio danRer7 61
## 9 Drosophila melanogaster dm3 502
## 10 Gallus gallus galGal3 32
## 11 Gallus gallus galGal4 15
## 12 Homo sapiens hg18 2512
## 13 Homo sapiens hg19 6876
## 14 Homo sapiens hg38 43
## 15 Mus musculus mm10 214
## 16 Mus musculus mm8 507
## 17 Mus musculus mm9 6289
## 18 Monodelphis domestica monDom5 8
## 19 Pan troglodytes panTro3 48
## 20 Pan troglodytes panTro4 42
## 21 Macaca mulatta rheMac2 81
## 22 Macaca mulatta rheMac3 31
## 23 Rattus norvegicus rn5 3
## 24 Saccharomyces cerevisiae sacCer2 141
## 25 Saccharomyces cerevisiae sacCer3 227
## 26 Sus scrofa susScr2 17
## 27 Xenopus (Silurana) tropicalis xenTro3 3 比如说斑马鱼,文件也就几十个,你大可全部下载下来。
downloadGEObedFiles(genome="danRer7", destDir="danRer7")
而比如人鼠这些明显特种,实在太多,一般来说全部下载也不太现实。ChIPseeker可以给你列出信息,连pubmed ID都给出来了,也方便翻阅文献,如果simplify=FALSE的话,还会给出protocal和data processing等信息哦。
hg19 <- getGEOInfo(genome="hg19", simplify=TRUE)
head(hg19)
## series_id gsm organism
## 111 GSE16256 GSM521889 Homo sapiens
## 112 GSE16256 GSM521887 Homo sapiens
## 113 GSE16256 GSM521883 Homo sapiens
## 114 GSE16256 GSM1010966 Homo sapiens
## 115 GSE16256 GSM896166 Homo sapiens
## 116 GSE16256 GSM910577 Homo sapiens
## title
## 111 Reference Epigenome: ChIP-Seq Analysis of H3K27me3 in IMR90 Cells; renlab.H3K27me3.IMR90-02.01
## 112 Reference Epigenome: ChIP-Seq Analysis of H3K27ac in IMR90 Cells; renlab.H3K27ac.IMR90-03.01
## 113 Reference Epigenome: ChIP-Seq Analysis of H3K14ac in IMR90 Cells; renlab.H3K14ac.IMR90-02.01
## 114 polyA RNA sequencing of STL003 Pancreas Cultured Cells; polyA-RNA-seq_STL003PA_r1a
## 115 Reference Epigenome: ChIP-Seq Analysis of H4K8ac in hESC H1 Cells; renlab.H4K8ac.hESC.H1.01.01
## 116 Reference Epigenome: ChIP-Seq Analysis of H3K4me1 in Human Spleen Tissue; renlab.H3K4me1.STL003SX.01.01
## supplementary_file
## 111 ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM521nnn/GSM521889/suppl/GSM521889_UCSD.IMR90.H3K27me3.SK05.bed.gz
## 112 ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM521nnn/GSM521887/suppl/GSM521887_UCSD.IMR90.H3K27ac.YL58.bed.gz
## 113 ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM521nnn/GSM521883/suppl/GSM521883_UCSD.IMR90.H3K14ac.SK17.bed.gz
## 114 ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM1010nnn/GSM1010966/suppl/GSM1010966_UCSD.Pancreas.mRNA-Seq.STL003.bed.gz
## 115 ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM896nnn/GSM896166/suppl/GSM896166_UCSD.H1.H4K8ac.AY17.bed.gz
## 116 ftp://ftp.ncbi.nlm.nih.gov/geo/samples/GSM910nnn/GSM910577/suppl/GSM910577_UCSD.Spleen.H3K4me1.STL003.bed.gz
## genomeVersion pubmed_id
## 111 hg19 19829295
## 112 hg19 19829295
## 113 hg19 19829295
## 114 hg19 19829295
## 115 hg19 19829295
## 116 hg19 19829295 然后经过你的过滤,你就可以选择性地下载部分文件来分析了。
gsm <- hg19$gsm[sample(nrow(hg19), 10)]
downloadGSMbedFiles(gsm, destDir="hg19") - 本文固定链接: https://maimengkong.com/image/1316.html
- 转载请注明: : 萌小白 2022年12月29日 于 卖萌控的博客 发表
- 百度已收录
