差异基因富集方法之Friends分析
大家好,我是阿琛。今天,我们来给大家介绍一种常见的拓展生信分析方法。在常规的⽣信文章中,我们往往能够得到⼀大堆的显著差异表达基因。然而,从⾥面挑选哪⼀个基因出来进行验证常常让我们感到困扰。 通常,我们会从这些特定的蛋白功能信息出发,根据差异基因的条⽬进⾏富集分析,查找与其功能相似或相关的蛋白质,关联、比较、量化,看看是否富集在某个GOterm或者KEGG通路当中。没错,这就是我们常说的功能富集分析。
但是,如果富集到的某个我们感兴趣的通路中基因数目依然比较多,那么如何从这⼀堆基因中挑选最重要的那个就是⼀个问题。哪些基因会比较重要呢?是基因的差异改变程度吗?然而,基因的Friends分析对此进行了重新定义。假设一个基因与通路上的其它基因都存在相互作用的话,那么这个基因就可能⽐较重要,可能是所谓的hub-gene。
下面,我们来看下如何根据基因信息进行Friends分析。
0. 期刊信息

1. R包和数据准备
1.1 R包安装与加载
这里,我们使用GOSemSim包进行差异蛋白的Friends分析;没错,这个包是由余叔在2010年发表于Bioinformatics杂志上,题目为 GOSemSim: an R package for measuring semantic similarity among GO terms and gene products,并将该包储存于Bioconductor网站上。大家在后续使用这个R包进行后续分析时记得引用该文章。

1.2 数据读取
接下来,我们将基因列表读取进来;通过str函数,可以看到,其中包含了8个不同基因的相关信息。

同时,使用as.character函数将数据框中ENTREZID号转换成字符串格式。

2. 计算相似性
首先,简单介绍一下,与大家熟悉的一样,常规的GO分析分为三种,分别为生物过程(biological process, BP),细胞组分(cellular component, CC),以及分子功能(molecular function, MF)。在此,我们同样从BP,CC和MF三个层面来进行分析。通过godata函数来构建来构建相应物种三个不同方面的GOSemSim所需的GO注释数据。
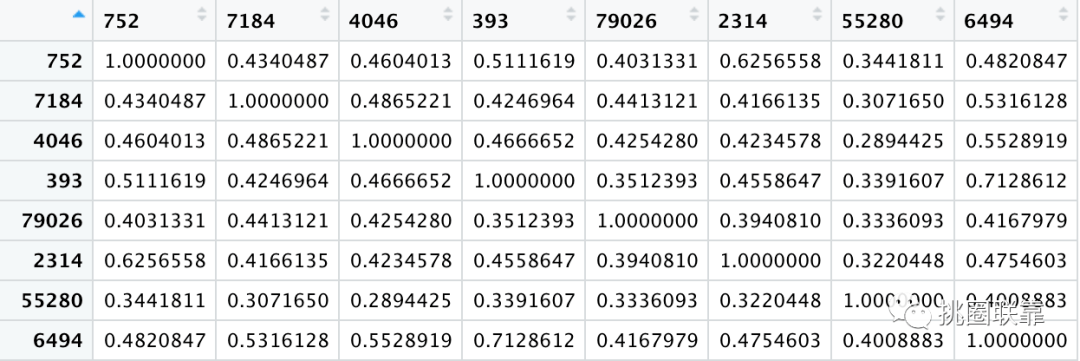
随后,根据前面得到的注释数据,使用mgeneSim函数,计算它们之间的语义相似度,最终得到它们之间的语义相似度和相应的GO语义相似度。
基于分析得到的相似度结果,我们进一步计算这些基因在BP,CC和MF层面的几何平均值,得到最终的评分,以用于综合评估该基因在三个层面的功能。
由于前面的分析是基于ENTREZID号进行的。为了便于后续的分析,我们将基因名从ENTREZID改为Gene Symbol。
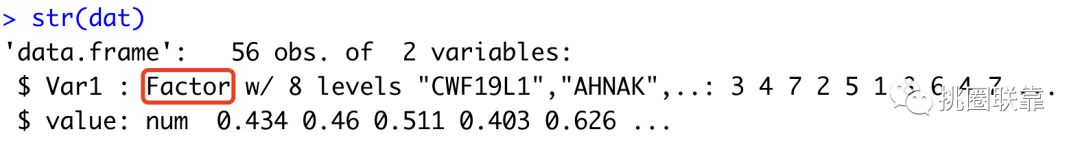
随后,进一步去除基因与本身基因之间的相关性,并使用melt函数将宽格式数据转化成长格式数据。

3. 绘制boxplot图
由于不同基因之间均可计算得到相似性评分,因此,我们使用aggregate函数需要计算所有评分的平均数。
同时,根据相似性评分的平均值,对其进行排序,并根据评分的高低,将基因名设置为因子(factor)格式。
最终,使用ggplot函数对Friends分析结果进行可视化展示。结果显示,基因SIPA1在其中发挥重要功能的基因,可能是重要的基因。

好啦,今天对内容就分享到这里了~大家可以根据文介绍的内容,给自己到文章进一步增添一张美图。
转自:解螺旋
- 本文固定链接: https://maimengkong.com/image/1154.html
- 转载请注明: : 萌小白 2022年8月20日 于 卖萌控的博客 发表
- 百度已收录
