导读
在农业科学中,为了提升作物农艺性状,经常会遇到将与性状相关的基因或位点在基因组上进行定位的需求,此时BSA作为一种简便又高效的分析方法便有了大显身手的机会。可是BSA究竟是怎样的一种研究方法呢,适用于什么群体呢?跟着小编了解一下吧!
什么是BSA?
BSA(Bulked segregation analysis)即混合分组分析,也称分离群体分组分析,是指利用目标性状存在极端表型差异的两个亲本构建分离群体,在子代分离群体中,选取两组表型差异极端的个体分别构建混合池 ,结合高通量测序技术对混合样本测序,比较两组群体在多态位点(SNP)的等位基因频率(AF)是否具有显著差异,定位与目标性状相关联的位点并对其进行注释,研究控制目标性状的基因及其分子机制。
相较于传统的遗传学研究方法(基因定位常用分析方法,小编已经安排上啦!),BSA最大的特点是不需要对群体中的所有个体进行基因分型,而是对挑选的个体按照性状进行混合分析,所以可以极大地降低研究的工作量和成本。
什么样本适合BSA分析?
既然BSA已经兼具了简便,准确、高性价比等优点,自然也有自己的小性子了,BSA分析对使用的群体有一定要求。
1、 人工构建的遗传群体(最常用来的是F2、BC、RIL)。通常来说,使用自然群体和遗传群体都可以进行BSA分析,但是考虑到遗传背景较复杂,可能导致定位结果不理想,所以不推荐使用自然群体进行BSA研究。
2、 亲本目标性状差异明显,其他性状差异随机分布,所构建分离群体两个混池之间目标性状有显著差异,非目标性状无明显差异。
3、 有合适的参考基因组信息。参考基因组组装的越好,信息越全,对于后续基因定位和候选区间的注释都会更加精确,可以锁定候选区间并估计候选区域的大小。没有组装到染色体级别的参考基因组,分析思路是一样的,但只能得到某个或某些scaffolds中的snp与性状相关,无法估计候选区间大小,甚至再组装结果差的情况下,无法判断基因的物理位置。
BSA有哪些分析方法?
1、SNP index及△SNP index
SNP-index作为主流的BSA定位的算法,最早在2013年被提出(Takagi)。它的基本原理是,构建子代分离群体,经过挑选极端性状构建混池后对SNP进行检测,对各混池进行等位基因频率分析,并与其中一个亲本进行比较。与此亲本不同的基因型所占的比例,即为该位点的SNP-index。从下图可以看到,两个位点的SNP-index分别为0.4和1。值得注意的是,这里的reference指的并不是我们进行重测序变异检测的参考基因组,而是我们构建群体所使用的亲本。这也是为什么进行SNP-index计算必须依赖于亲本测序数据的缘故。
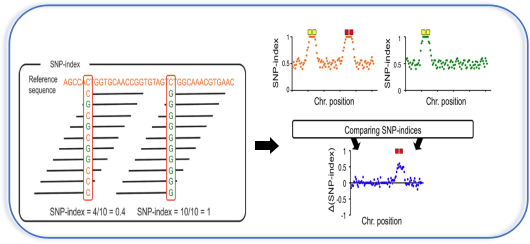
每个混池都得到一组SNP-index数据之后,两个混池相减(上图右),即得到了△SNP-index的结果,代表的是两个混池之间SNP基因型频率的差异。理论上说,不与性状相关的位点,△SNP-index的值应当在0左右,代表混池之间不存在差异;而QTL及其相连锁位置的SNP,△SNP-index值应当呈现较高的数值。△SNP index这种分析方法会存在因统计偏差造成的假阳性位点,这时我们可以通过计算滑窗内所有SNP的△SNP-index,来消除其影响,得到真正QTL所在的基因组区域。
2、 欧几里得距离(ED)
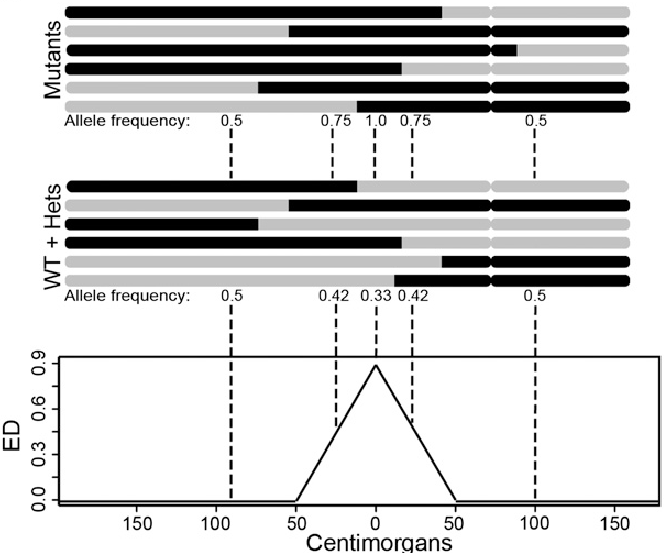
随着BSA技术的发展,SNP-index显示出了一定的局限性,比如亲本数据缺失,林木类较难构建分析群体,ED值的分析方法应运而生。在BSA和BSR中,欧几里得距离可以计算同一个位点上,两个混池之间的等位基因频率。两个极端性状子代混池只在控制性状的QTL及其连锁位点出现差异,所以通过各个位点欧几里得距离的计算,我们可以判断哪些位点更可能是控制对应性状的QTL。计算公式如下:
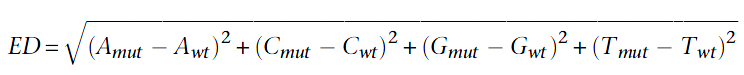
实际应用中,我们在BSA的两组混池之间可能会得到数十万甚至上百万个SNP,有的SNP可能实际与性状无关,但因为抽样偏差,导致计算得到的ED值很高,为了排除统计异常值,我们通常会采用滑窗对在一个窗口内所有位点的ED值进行拟合,消除抽样偏差产生的假阳性结果。而在BSA定位区间计算过程中,会对ED值采取乘方处理,放大ED值的差异,使定位区间更加明显。
3、 Gradedpool-seq(Ridit检验)
Gradedpool-seq的概念在2019年由韩斌和黄学辉课题组提出并发表于Nature Communication(Wang et al., 2019)。这种方法与常规BSA类似的是,它也是基于性状分离群体中按照性状选择子代个体构成混池(通常加上亲本)进行测序,并进行QTL定位的方式。Ridit是relative to an identified distribution unit一词的缩写,它是一种非参数检验分析方法,用于按等级分组资料的比较。而对于多个混池测序数据,Ridit检验会对每个位点的等位基因频率进行计算,判断其是否显著偏离标准分布,得到一个p值。换言之,这个位点的p值越小,即代表这个位点与性状相关联的可能性越高(与GWAS关联方法类似)。
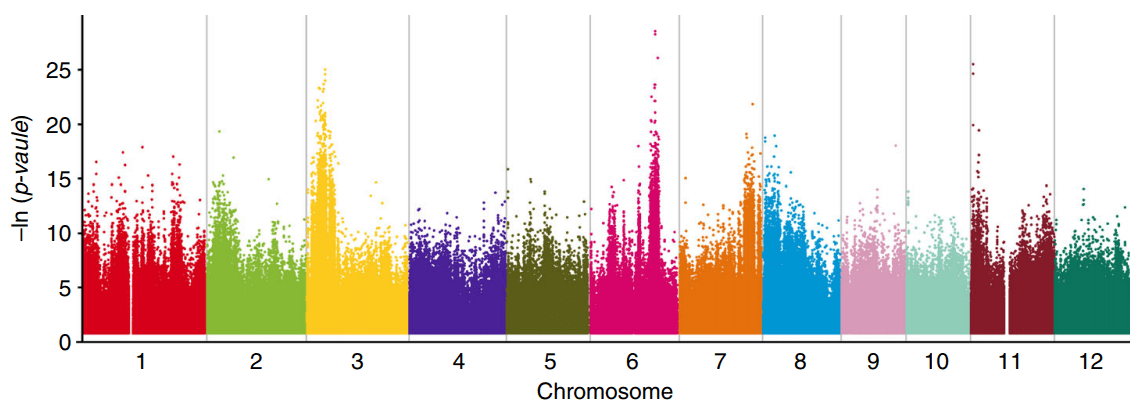
由于在BSA项目中Ridit检验的对象只有2-4个混池,基因型数据较少,所以当Ridit检验的结果用曼哈顿图的形式展现出来,其噪音非常强烈,很难从中直观地判断我们的候选区间的位置。研究者们选取一定大小的窗口,并且将窗口内的SNP位点进行统计,计算p值低于阈值的位点所占的比例。一般经过这种滑窗降噪处理,其关联区间也就显现出来了。
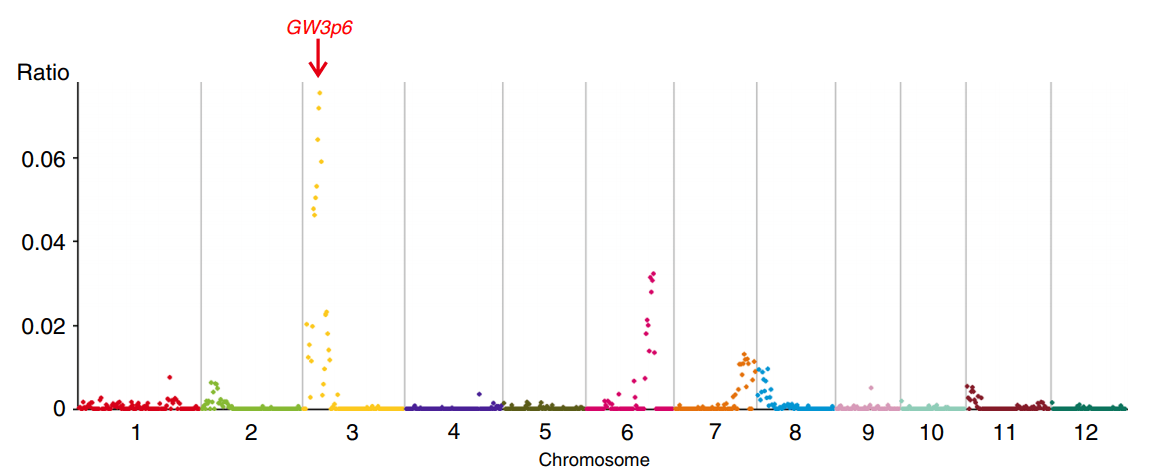
好啦,唠叨了这么多,不知道大家是不是有所收获呢?对于这种即简便又实用的小可爱,是不是难以拒绝呢?区间定位到了,后续如何进行精细定位与验证呢?请听小编下回分解啦!
参考文献:
1.Hill JT,et al. MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 2013, 23(4):687-97
2.Takagi H, Abe A,Yoshida K, et al. QTL‐seq: rapid mapping of quantitative trait loci in riceby wholegenome resequencing of DNA from two bulked populations[J]. Plant Journal,2013,74(1):174-83.
3.Wang, C., Tang, S., Zhan, Q. et al. Dissecting a heterotic gene through GradedPool-Seq mapping informs a rice-improvement strategy. Nat Commun 10, 2982 (2019).- 本文固定链接: https://maimengkong.com/1201.html
- 转载请注明: : 萌小白 2022年10月1日 于 卖萌控的博客 发表
- 百度已收录
