常规分析找不到差异基因?GSEA分析了解一下
在上一期中,我们云平台的生信工程师为大家带来了一款强大的分析工具GSEA,很多老师觉得那篇稿子三分钟绘制一张优美的GSEA图 | 云平台看完后仍然意犹未尽。不过大家请不要着急,今天会带领大家对一些概念重新温习一遍。
在引出今天的主角GSEA分析之前,我们先来谈一谈大家在挖掘转录组数据中,可能会遇到的一些共性问题——差异分析拿到了一大堆基因无从下手,或者拿不到自己要研究的那个差异基因进行下游的研究。
究竟是测序公司分析的有问题还是自己的实验设计有问题呢?别急,这个问题我们下面会慢慢聊。
常规差异分析存在的问题
当你想要研究癌症组织和癌旁组织究竟哪些基因有差异表达时,你会把组织寄送到测序公司提取RNA进行转录组测序。
测序公司标准的差异分析默认不考虑通路,直接进行负二项式分布法、F检验、2×2卡方分析或T-test等各种你听过的或没有听过的统计学方法,对所有有表达的基因进行差异分析。这种方法简单粗暴,每个基因在每组差异分析中都会得到一个p value和fold change值。
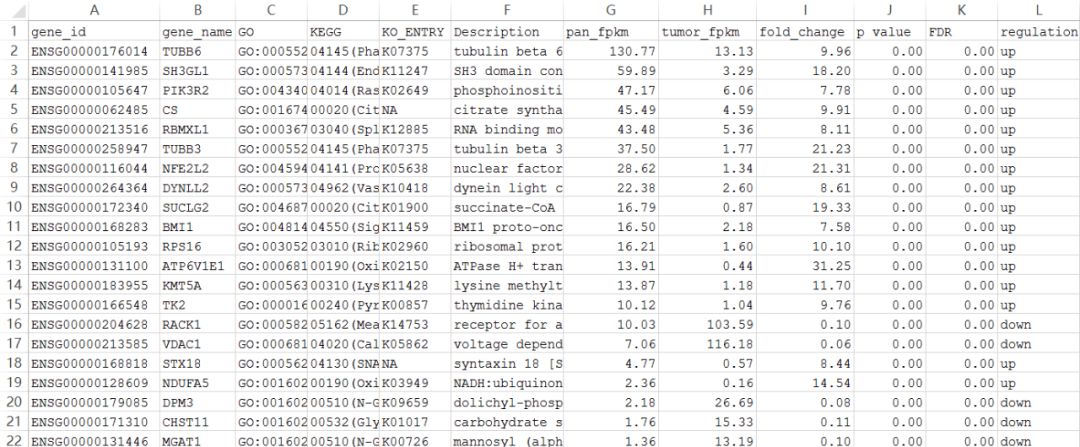
最后你会拿到一个测序公司发给你的差异分析表格。从上图中,我们可以看到,这21个基因基本上差异倍数非常大,且p value都接近于0。通常我们认为,从纯统计学角度来看,只要p<0.05且fold change>2或<0.5,这个基因就是有差异的。
接下来你会面临两种常见的情况:差异基因过多,有将近1000个基因……或差异基因过少只有不到20个。这时候你该怎么办?
假如差异基因有1000个,这时候并不是所有差异基因都是你要研究的对象。你会选择预设的信号通路或感兴趣的候选基因进一步缩小范围。比如把差异基因中与Wnt通路相关的基因都单独拿出来,或者把自己预先感兴趣的40个候选基因与这1000个差异基因取交集后看看还剩下哪些。
不过当你手中只有19个差异基因时,你扫了一圈发现这19个基因里居然全都有没有你要的那些基因!
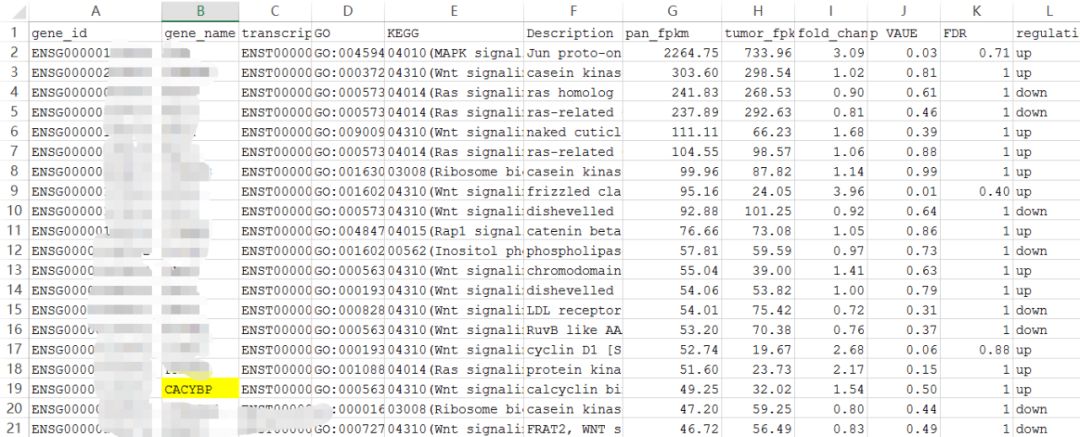
举个例子如上图所示,最郁闷的事情来了,无论是差异基因过多还是过少,你最最感兴趣的CACYBP并不在当中,而这个基因确实是你这项研究中十分关键的分子。那这批数据没办法用了吗?
这时候你需要问自己一个问题,假设某个你很感兴趣的基因在测序结果中,差异倍数只有1.6倍,p value≈0.09,那么这个基因究竟是要继续深入做还是放弃?这批数据还能用吗?
如果你回答是这批数据不能用了,那么你就不必往下看了(开玩笑)。事实上许多基因在差异倍数不足2倍时,仍然在调控过程中发挥巨大的作用。下面我们就会对传统的数据方法进行讨论,从而引出一些标准差异分析的一些弊病以及GSEA分析所具有的独特优势。
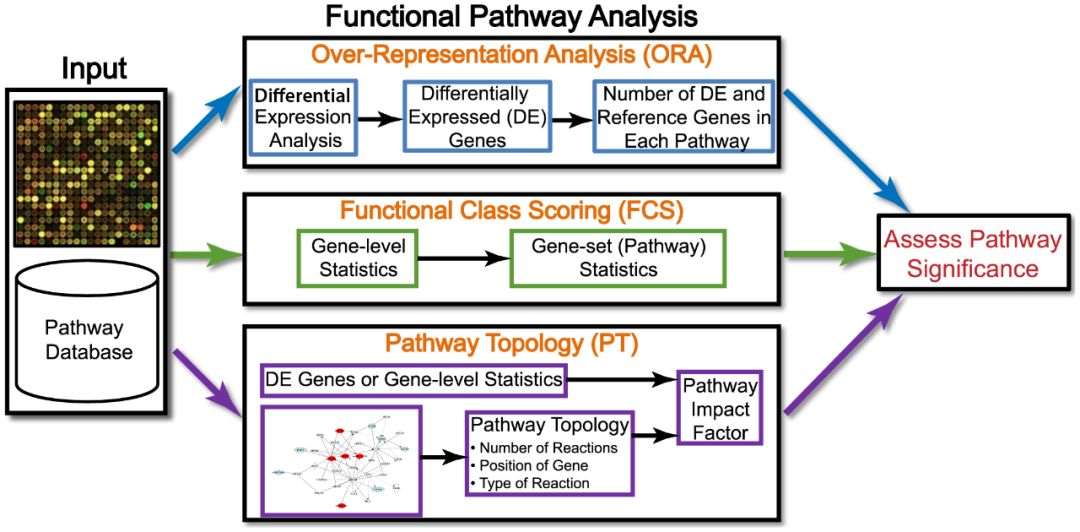
(该内容引用自【高通量测序数据处理学习记录(三):Pathway Analysis及GSEA】https://www.jianshu.com/p/be1211dce097)
1. ORA:Over-representation analysis
曾经一段时间,基因芯片microarray技术的风靡产生了对下游分析的极大需求, over-representation analysis (ORA) 应运而生,从一系列基因里根据阈值提取出部分基因进行显著性分析,也就是统计学常见的“2X2 交叉表格法”,对每个pathway进行统计分析,常见tests都建立在 hypergeometric distribution 、chi-square、binomial distribution 等方法上。
ORA分析缺陷:
① 只考虑了差异基因列表,并不考虑差异基因的表达情况
② 只检验了通过设定阈值筛选标准的基因,对差异微弱的基因并未考虑,但实际上会造成bias
③ 每个基因会作为独立存在事件考虑,未加入相互影响干扰因素
④ Pathway也是作为独立存在事件考虑
我们日常分析中最常见的GO/KEGG 分析就是基于这种原理,虽然老旧但实用
2. FCS:Functional Class Scoring Approaches
FCA 的推测设想认为虽然强烈的单个基因的改变可以影响到pathways,但是微弱的相互协同的功能相关基因的变化也可以拥有这种影响,所以这种方法的输入数据是一个基因水平的统计数据(标准化后食用更佳),随后把gene-level的数据输入到pathway-level进行统计,现有方法包括Kolmogorov-Smirnov statistic, sum, mean, or median of gene-level statistic, the Wilcoxon rank sum, and the maxmean statistic 等,最后再做一个显著性检验。
相对于ORA,FCS完善了三个缺陷:
①不需要人为的阈值确定差异基因list
②FCS使用所有可用的表达水平进行分析
③FCS考虑了基因相互间的变化,解释了基因变化与pathway之间的依赖性
FCS分析的缺陷:
①类似于ORA,pathway之间的分析依旧是彼此独立的(此种原因可以解释为单个基因同时存有多种功能,在多个pathway中发挥作用,overlap过多的pathway就会相互干扰)
②使用rank的方式纵然有着很多优点,但是忽略了单个基因的变化幅度,也就是权重
3. PT:Pathway Topology -Based Approaches
因为ORA和FCS只考虑了基因而未利用额外的数据信息所以天然的存有着分析短板。PT 就是尝试利用额外的信息进行统筹分析,但是它其实和FCS的分析过程是没有差别的,唯一的区别在于在进行gene-level statistics的时候使用pathway topology方法。
Rahnenfuhrer et al.推出的ScorePAGE,通过计算相关和协方差的方式来得到类似于FCS的pathway-level的结果,但是又综合考虑了两组gene list之间需要connect的难度从而进行给分,而不是像FCS分配统一权重。
PT分析缺陷:
① PT-based的方法千差万别,结论也千差万别,很难界定结果的准确性
② 精确的分析结果依赖于数据库的信息准确性,但是细胞特异性的基因表达数据目前还非常不完善,这也是卡住方法开发的门槛
③ 相关分析无法考虑动态变化,毕竟生物系统是一个不断协调变化的过程
一般常规的差异分析,往往从纯统计学角度集中关注几个显著上下调的基因,我们称之为TOP基因。p value<0.05且fold change>2的这种一刀切的粗暴做法,很容易把不参与重要生物学功能但有统计学意义的TOP基因放在首位,而一些表达差异不显著却有重要生物学意义的基因容易被误差和遗漏。
想想早期的全血样本中,是不是有大量的肌红蛋白基因超高表达?亦或是血清样本中,有大量的IgG蛋白让你头疼不已。这些基因或蛋白虽然很重要,但是容易对低丰度的一些基因或蛋白产生巨大的干扰。
又比如处于调控上游的转录因子往往只要有不到2倍的变化,就会引起下游基因乃至蛋白高达几十倍的变化。蛋白的细微变化又会引起代谢产物几百倍的变化。
处于调控网络中的一些基因可以被看作是一个整体,常规的差异分析过分强调个体,导致遗漏信息。当研究相同的生物作用通路时,两项研究中具有统计学意义的基因列表可能会很少有重叠。
那么常规的GO和KEGG富集分析是如何做的呢?首先依旧是把所有的差异基因都列出来,比如前期通过测序拿到了400个差异基因。这些差异基因都会对应的GO和KEGG注释。
假设其中300个基因都在Wnt通路上有注释,270个基因在MAPK通路上有注释(可能跟Wnt通路有交集),20个基因在Akt通路上有注释……诸如此类。根据负二项式分布计算来看,根据通路注释到的基因数量而言,Wnt通路显然是差异极显著。
但是问题来了,假设这300个基因fpkm也就是表达量没有超过0.01的,这种极端情况出现后我们就认为哪怕300个基因有差异但表达量极低,无法行使重要的生物学功能。而有些只有20多个基因富集到Akt通路上,但是表达量很高差异也很大。那么这种GO和KEGG富集分析结果将不会有特别大的参考意义。
GSEA概念及优势
GSEA全称是Gene Set Enrichment Analysis,也叫基因集富集分析。其基本思想是使用预定义的基因集,将基因按照在两类样本中的差异表达程度排序,然后检验预先设定的基因集合(Gene Set)是否在这个排序表的顶端或者底端富集。
GSEA分析最关键的就是基因集(Gene Set)。这个基因集通常是人为事先进行预设及定义的,在GSEA的官网(http://software.broadinstitute.org/gsea) 就可以进行下载。包括免疫相关基因等都包含在内一应俱全。如果对这些网上整理好的基因集合不感兴趣,还可以人为进行手动自定义。如最近m6A甲基化比较火热,许多老师会把自己感兴趣的10多种甲基化酶和下游某几个明星分子整合到一起,形成一个自定义基因集合。
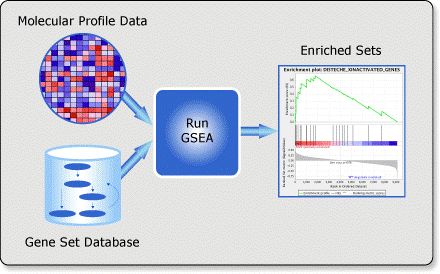
上图就是简单说明了GSEA分析的大致流程。左上方是你的基因表达谱数据,这边以热图的形式作为展示,你输入的数据是所有样本所有的基因表达谱。左下方是预设的基因集合。准备好两个文件后,再进行GSEA分析,最后获得富集显著的一些基因。
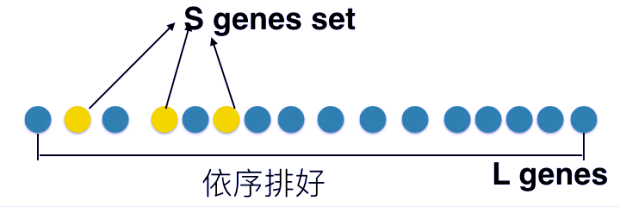
GSEA分析的原理
具体原理如上图所示,在给定一个排序的基因表L和一个预先定义的基因集S(比如编码某个代谢通路的产物的基因,基因组上物理位置相近的基因,或同一GO注释下的基因),GSEA的目的是判断S里面的成员s在L里面是随机分布还是主要聚集在L的顶部或底部。这些基因排序的依据是其在不同表型状态下的表达差异,若研究的基因集S的成员显著聚集在L的顶部或底部,则说明此基因集成员对表型的差异有贡献,也是我们关注的基因集。
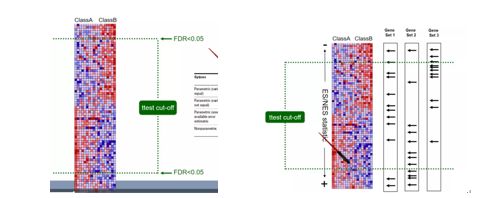
接下来,我们来更加直观看一下常规差异分析和GSEA分析究竟有何不同。从上图我们可以看到,左图是典型的差异分析即双边检验只要显著差异表达的基因就行了。而右边我们可以看到,由于基因集合的引入即图中显示的Gene Set1、Gene Set2 Gene Set3等,我们可以更加直观看到这种优化过后的结果更符合我们的预期。
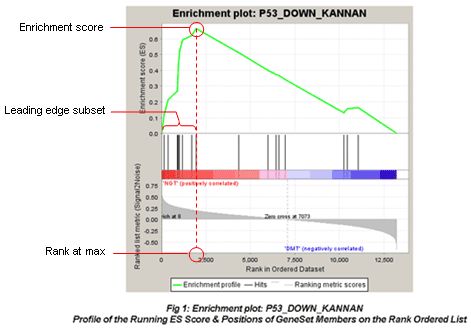
至于GSEA分析后得到的ES峰图,许多老师说不太看得懂,这边我们会进行小小的解释。
①图最上面部分展示的是ES值计算过程,从左至右每到一个基因,计算出一个ES值,连成线。最高峰为富集得分(ES)。在最左侧或最右侧有一个特别明显的峰的基因集通常是感兴趣的基因集。
②图中间部分每一条线代表基因集中的一个基因,及其在基因列表中的排序位置,即,本次gene sets里面的基因在本基因的位置。
③最下面部分展示的是基因与表型关联的矩阵,红色为与第一个表型(MUT)正相关,在MUT中表达高,蓝色与第二个表型(WT)正相关,在WT中表达高。
④Leading-edge subset:定义其中对Enrichment score贡献最大的基因为核心基因。若富集得分为正值,则是峰左侧的基因;若富集得分为负值,则是峰右侧的基因。
⑤FDR GSEA默认提供所有的分析结果,并且设定FDR<0.25为可信的富集,最可能获得有功能研究价值的结果。但如果样品数目少,而且选择了gene_set作为Permumation type则需要使用更为严格的标准,比如FDR<0.05。
⑥如果gene set里面的基因集中在2万个基因的前面部分,就是在case里面富集,如果集中在后面部分,就是在control里面富集着。
需要再次强调的是,基因集合富集分析检测的是基因集合(也就是一堆基因)而不是单个基因的表达变化,因此这种分析结果可以包含所有基因更多细微的表达变化,预期得到更为理想的结果。
我们假设上面提到的癌症组织和癌旁组织提供得到2000个表达的基因,而且还关注Wnt通路相关的基因。这时候我们在准备文件的时,除了癌症癌旁组织的2000个基因表达谱信息外,还需要输入额外的Wnt通路相关的基因集合(网上可以直接下载)。把两个文件作为输入文件进行GSEA分析后,会告诉你某几十个基因显著富集(也就是差异分析中显著差异)。
我们再来重复一下GSEA分析的几个特点:
①分析出来的差异或者说是富集的结果是基因集合而不是单个基因;
②将基因与预定义的基因集进行比较;
③可以将被统计学误杀的重要基因重新“补救”回来
GSEA官方预设的基因集合介绍
GSEA分析最关键的就是基因集(Gene Set)。这个基因集通常是人为事先进行预设及定义的,在GSEA的官网(http://software.broadinstitute.org/gsea) 我们可以看到一共有包括H和C1-C7在内的八个大类,具体介绍如下
①H: hallmark gene sets (效应)特征基因集合,共50组


官网介绍说这些基因集合代表着特定明确的生物学过程,并呈现出连续表达的状态。通过特定的算法对C1-C6中可能存在Overlap基因进行去冗余,得到了50个高质量的hallmarks。

从上图中我们看到,这50个hallmarks中包括了大量的经典基因集合,如脂肪形成、自噬、DNA修复、低氧胁迫、心肌细胞再生、炎症应答、干扰素α应答、干扰素β应答。信号通路基因集合还包括了hedgehog信号通路、Notch信号通路、白介素6介导JAK-STAT3信号通路、TGF-β信号通路等。
这些经典的基因集合,将会在后期大大缩短老师们筛选数据的时间。即便不进行GSEA分析,这些经典的基因集合也会为大家在后期的数据挖掘提供极大的便利。
②C1: positional gene sets位置基因集合,根据染色体位置,共326个。


这些基因集合通常根据染色体的位置而定。对于一些做GWAS+转录组研究的老师来说,是非常不错的选择。
③C2: curated gene sets:(专家)共识基因集合,基于通路、文献等已有的知识储备体系整理而成。
C2这个基因集合里,包含了庞大的信息量。如果细分的话还可以再细分为化学及遗传扰动CGP(Chemical and genetic perturbations)和经典信号通路CP(Canonical pathway)。
我们所熟知的KEGG pathway、BioCarta、Matrisome Project、Pathway Interaction Database、Reactome、Signaling Gateway等都包含在内。另外一些 来自工业界的科学家们也整理了大量的信息,包括Sigma化学、sabiosciences、安捷伦生命科学部、昂飞Affymetrix等也参与了大量的工作。


这些手动整理的基因集合,将会在后期研究中提供大大的便利。从上图中我们可以看到,具体可以细分到每个数据库都可以focus到非常narrow的点。
④C3: motif gene sets:模式基因集合,主要包括microRNA和转录因子靶基因两部分


这些基因集合目的非常明确,就是找特定的靶向蛋白或靶向基因。无论是miRNA还是转录因子,数据中都收录了大量分基因集合信息,总计达800多个sets。

从上图中可以看到,无论是miRNA还是转录因子本身,都可以与大量的基因相结合。这块工作信息量巨大,非常非常适合有特定研究目的的老师,如miR-203的靶基因研究等。
⑤C4: computational gene sets:计算基因集合,通过挖掘癌症相关芯片数据定义的基因集合

⑥C5: GO gene sets:Gene Ontology 基因本体论,包括BP(生物学过程biological process,细胞原件cellular component和分子功能molecular function三部分)

大名鼎鼎的Gene Ontology简称GO,是所有生信分析中高频出现的数据库注释之一。这边就不再做更多的介绍了。关于GO数据库的详细信息,请点击 http://geneontology.org/ 查询。
⑦C6: oncogenic signatures:癌症特征基因集合,大部分来源于NCBI GEO 未发表芯片数据

⑧C7: immunologic signatures: 免疫相关基因集合


对于从事免疫学研究的老师来说,这个C7的基因集合无论是新手还是老鸟,都非常非常重要。C7基因集合庞大到多大近5000个sets。
这些与免疫相关的基因信息都是由人工手动整理完成,对于存储在NCBI的GEO数据库所有关于免疫学研究的芯片数据和测序数据进行整理分析并得到了高质量的基因集合。
联川生物云平台GSEA分析实战解析(部分关键信息已经做了隐藏)
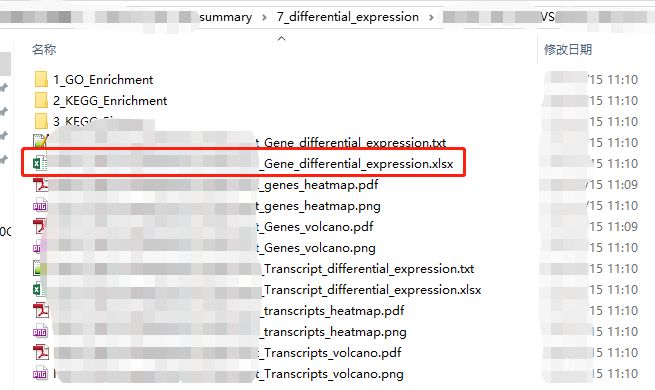
第一步,当然是拿到我们的表达谱数据。如上图所示,以转录组为例。在我们差异分析结果中,你需要在差异表格所在的文件夹(通常以differential_expression作为关键词)里,找到以*_Gene_differential_expression.xlsx结尾的文件。
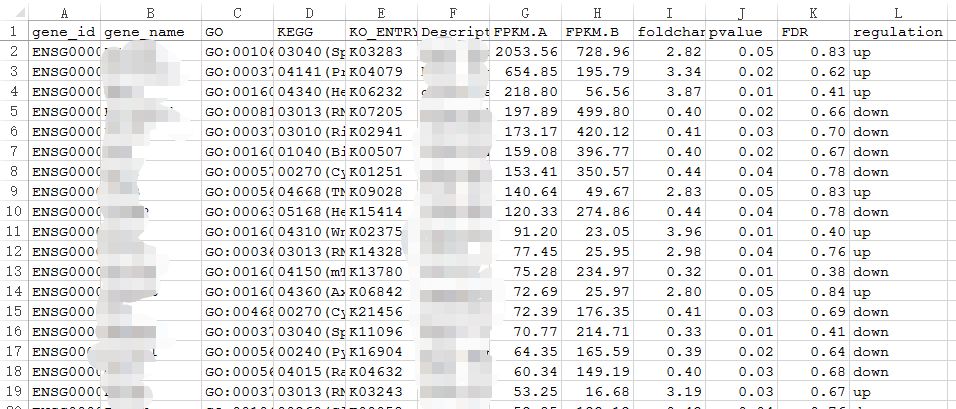
第二步,打开*_Gene_differential_expression.xlsx文件,你会发现有5000多个基因有表达,其中差异个数280个。但是这280个中并没有发现想要的一些差异基因。

第三步,把*_Gene_differential_expression.xlsx中的核心信息整理出来,即基因名加表达谱信息。所以最后我们就得到了上图所示的新文件。你会发现里面只有三列,基因名加样本A和样本B的表达谱信息,并保存为test.xlsx。
第四步,打开联川生物云平台,登录后点击GSEA富集分析。
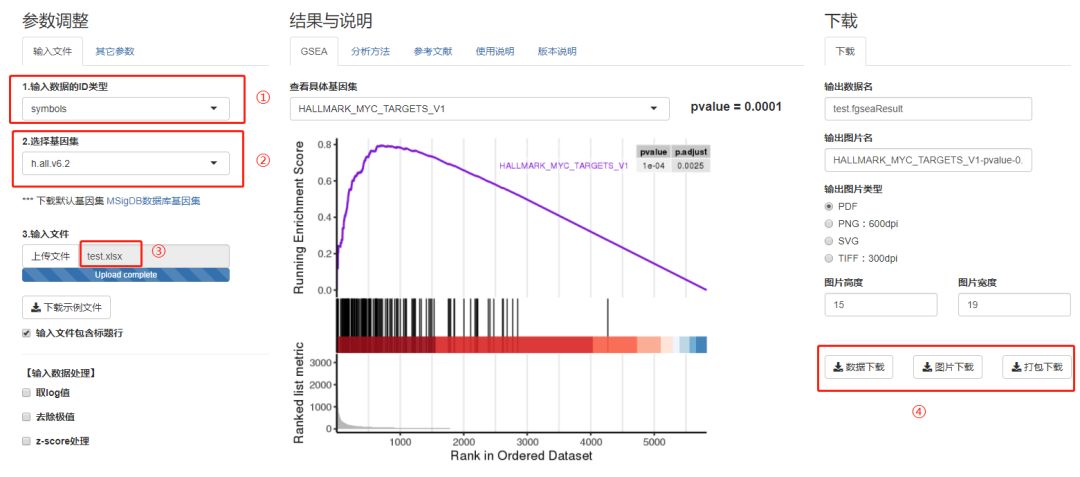
第五步,我们上传的数据test.xlsx。需要注意的是:①我们要选择symbols,如果是entrez那么最后的富集到的基因名都是数字;② 基因集这边我们选择H hallmarks,这边老师可以根据我们前面的介绍选择自己感兴趣的基因集,如对免疫感兴趣可以选择c6等;③ 上传文件最好格式要符合要求;④ 这边既可以单独下载图片和表格,也可以选择打包下载。
第六步,我们在5个富集的通路上,找自己这批数据中的基因集合信息。最后一列,就是每个通路对应在本次测序数据中对应富集基因。具体操作请参考三分钟绘制一张优美的PCoA图 | 云平台- 本文固定链接: https://maimengkong.com/zu/1249.html
- 转载请注明: : 萌小白 2022年10月6日 于 卖萌控的博客 发表
- 百度已收录
