上上上期靠谱er在连锁分析和关联分析的稿子( 关联分析和连锁分析 | 群体遗传专题 )最后留了个尾巴,这次就把这个尾巴续上,讲讲连锁分析和关联分析的文献案例。
总览
玉米籽粒相关性状和产量直接相关。本文通过关联群体和双亲杂交群体解析三个籽粒性状(籽粒长度,籽粒快递,籽粒厚度)的遗传基础。在四种环境下共检测到21个和以上三个性状关联的位点。另又在7种环境下通过B73和Mo17构建的DH系群体共定位到50个QTL,其中8个能至少在3种环境下重复检测到。联合连锁和关联分析的结果,发现在18个QTL区间内有56个显著关联的SNP。基于SNP的关联程度(P值),SNP的效应稳定性和共定位的SNP三个指标,作者共鉴定到了73个调控籽粒发育的候选基因。此外,在位于共定位的SNP的LD block里发现有7个miRNAs,其中一个zma-miR164e,被证明能够剪切拟南芥的CUC1,CUC2和NAC6的mRNAs.过表达该miRNA使以上三个基因下调,同时影响拟南芥荚中籽粒形成,但是能增加分枝数量。本文的发现为种子发育提供了新见解,也有利于基于分子标记辅助选择(MAS)的玉米高产性状改良。
材料和方法
关联分析群体:310个来自于中国西南部育种program的玉米自交系,完全随机区组实验设计,两个生物学重复。每个plot包含14株,3m长,0.75米宽。每个自交系种植一行。2016年时候种植于西双版纳景洪,四川洪雅,四川雅安三个地点。BLUP处理以上三个地点的表型后形成另一个供GWAS关联的表型。每个自交系每个重复随机取5个自交后的穗,每个穗中部选择10个籽粒测量粒长,粒宽和粒厚。最终以两个试验重复的平均值作为关联分析的表型。
连锁分析群体:265个B73和Mo17构建的DH系群体用来进行QTL定位。该群体种植于6种不同的环境下,包括四川崇州(2016,2017),四川温江(2016,2017),西双版纳景洪(2016)和新疆昌吉(2017)。粒长和粒宽表型均进行了考察,粒厚只在以上3种环境下进行了考察。具体种质情况和关联群体一致。
关联分析方法:基于已经发表的文章中基于芯片的SNP (Zhang et al., 2016),先做如下的过滤:缺失率>5%;杂合度>20%,MAF<0.05. 然后过滤掉非二等位的SNP panel(39354个SNP)后用于LD计算和GWAS分析。群体结构分析的SNP 从以上的SNP panel中随机选择5000个,基于贝叶斯马尔科夫链蒙特卡洛法进行样品分群。R语言中的PCA降维。最优亚群用来计算Q矩阵,SpAGeDi用来计算亲缘关系矩阵,TASSEL5.0 用来计算LD和GWAS分析。GWAS分析采用CMLM、MLM和FarmCPU三种模型来平衡假阳性和假阴性结果。SNP显著性阈值先采用simpleM法估计有效SNP数量,然后计算族系误差率(FWE)。比Bonferroni校正的阈值要严格……
连锁分析方法:连锁分析材料已经发表的含有6618个bin的高密度遗传图谱,平均图距为0.48 cM。基于Windows QTL Cartgrapher v2.5 的CIM法进行QTL定位。阈值固定为LOD=2.5.
miRNA的靶基因预测:zma-miR164e在拟南芥中的靶基因预测在网站进行:http://plantgrn.noble.org/psRNATarget/。选择前三个miss match的基因作为潜在的靶基因。
其他载体构建、转基因方法不再赘述。
结果分析
1. 基因定位的paper一般就是先描述表型的结果,本文非常简单的描述了两种群体的表型变异情况、广义遗传力,另外还分析了三个表型之间的相关性。完了下了结论:本文的表型数据比较可靠,可用于下游的关联分析和连锁分析。非常常规的操作。由于数据量较大,表格均放入了附表中,这部分作者只拍了一张图片。
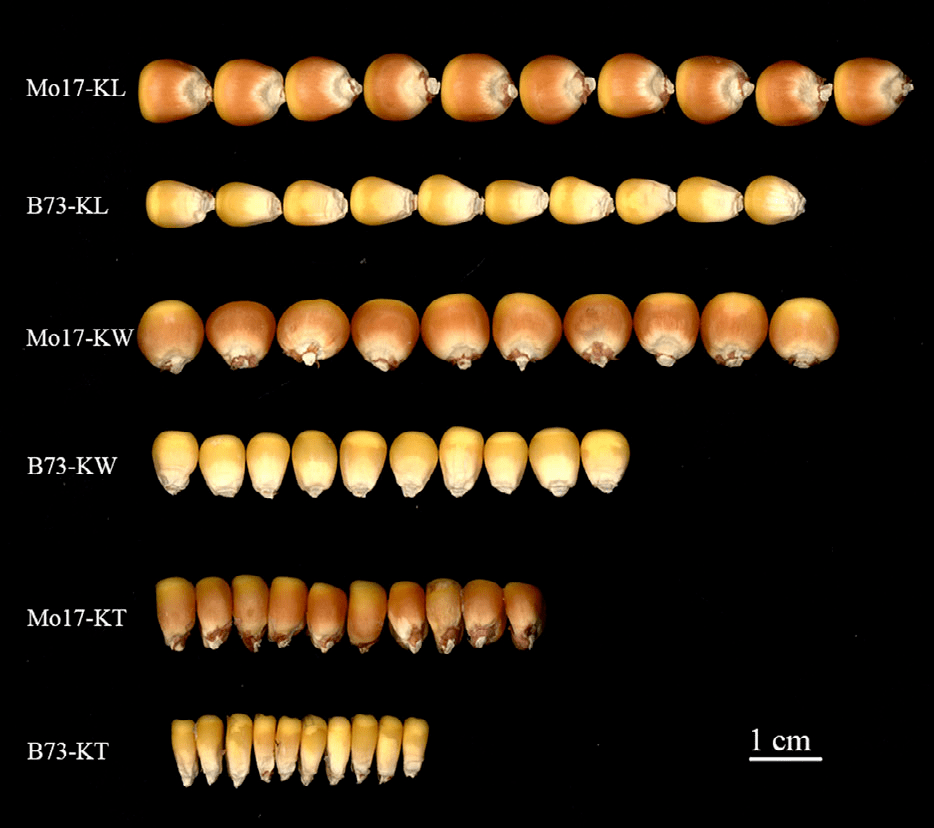
图1 典型表型展现
2. 关联群体的结果描述。首先是群体结构和LD分析。由于本文的材料只来自与西南部,只分成了4个亚群。LD衰减为220 kb,远大于玉米的5-10 kb的LD decay。以上的结果说明本文的自交系遗传背景较窄,带来的后果是定位的分辨率稍低,但是群体结构相对不复杂,也降低了定位的假阳性概率。三种模型的GWAS结果发现,GAPIT关联到2个SNP,TASSEL关联到1个SNP,FarmCPU关联到20个(有可能选择3个模型是被逼的,因为前两个位点太少了……)。1号染色体上的2个位点能同时被2个模型支持。这些SNP中,4个位于基因间区,17个位于基因内。这对应的17个基因中,有控制籽粒发育的基因EMBRYO DEFECTIVE 2733的同源基因,也有E3泛素连接酶基因HRD1A和其他相关基因。由于关联到的SNP很少,为了能比较不同环境下的关联结果,作者降低了阈值(1X10 4)。此番操作后果然发现了13个能在不同环境下均能检测到的SNP位点。这里边2个和籽粒长度相关的SNP能在所有的环境中都能检测到,且能被所有的模型检测到。这13个基因中作者筛选到9个候选基因,其中,包含了和籽粒发育相关的RAD23C、泛素活化酶E12的同源基因。
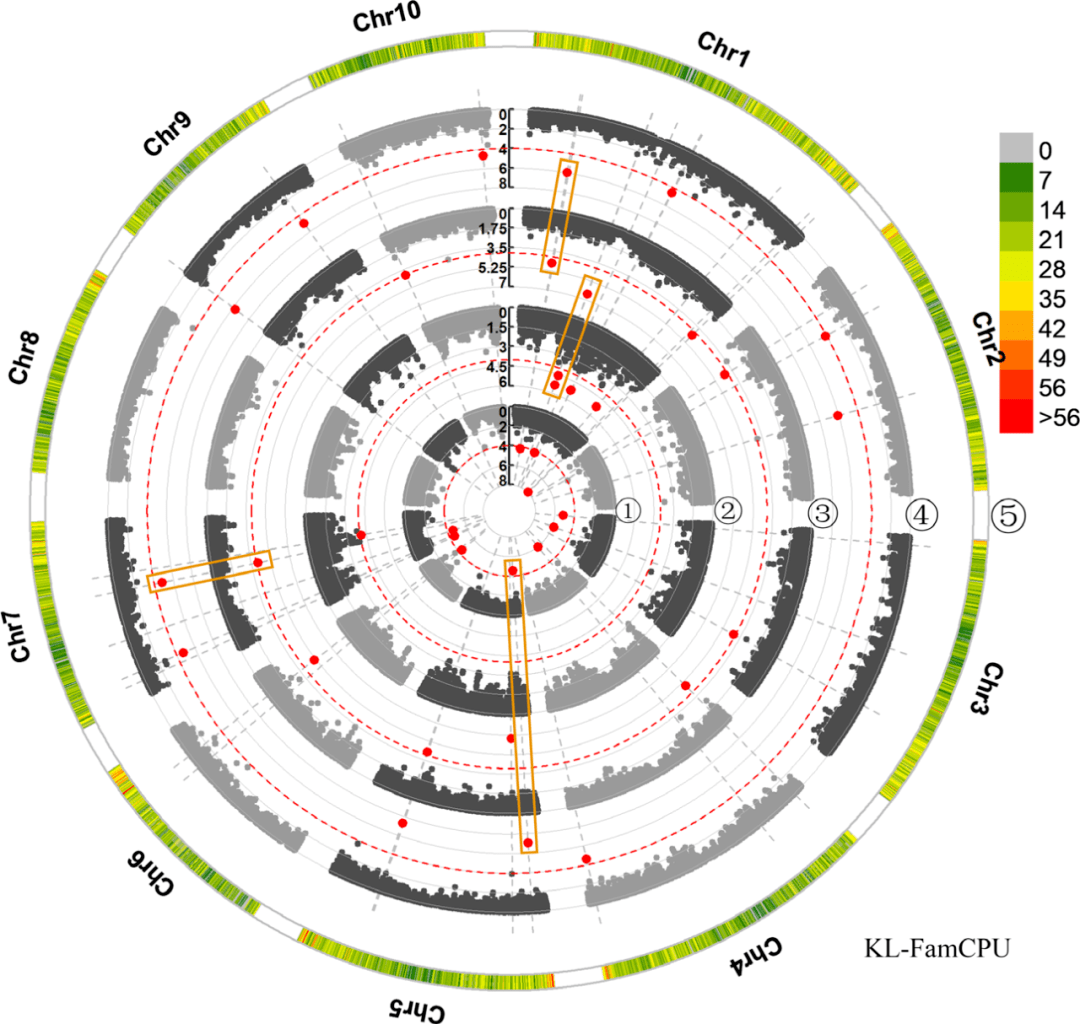
图2 GWAS的定位结果
3. QTL定位结果描述。基于单个环境的QTL分析,总共有50个QTL被定位到。定位的区间从0.2 Mb到24.8Mb,平均3.82MB(这就是265个DH系的定位分辨率)。PVE从3.48%到15.04%(有超过10%的就不错)。为了比较不同环境的QTL定位,作者做了如下的规定:只要不同环境下有overlap的QTL,就被视为一个QTL。这样,作者发现有18个QTL至少能同时出现在2种环境中。其他扒拉扒拉就不说了。
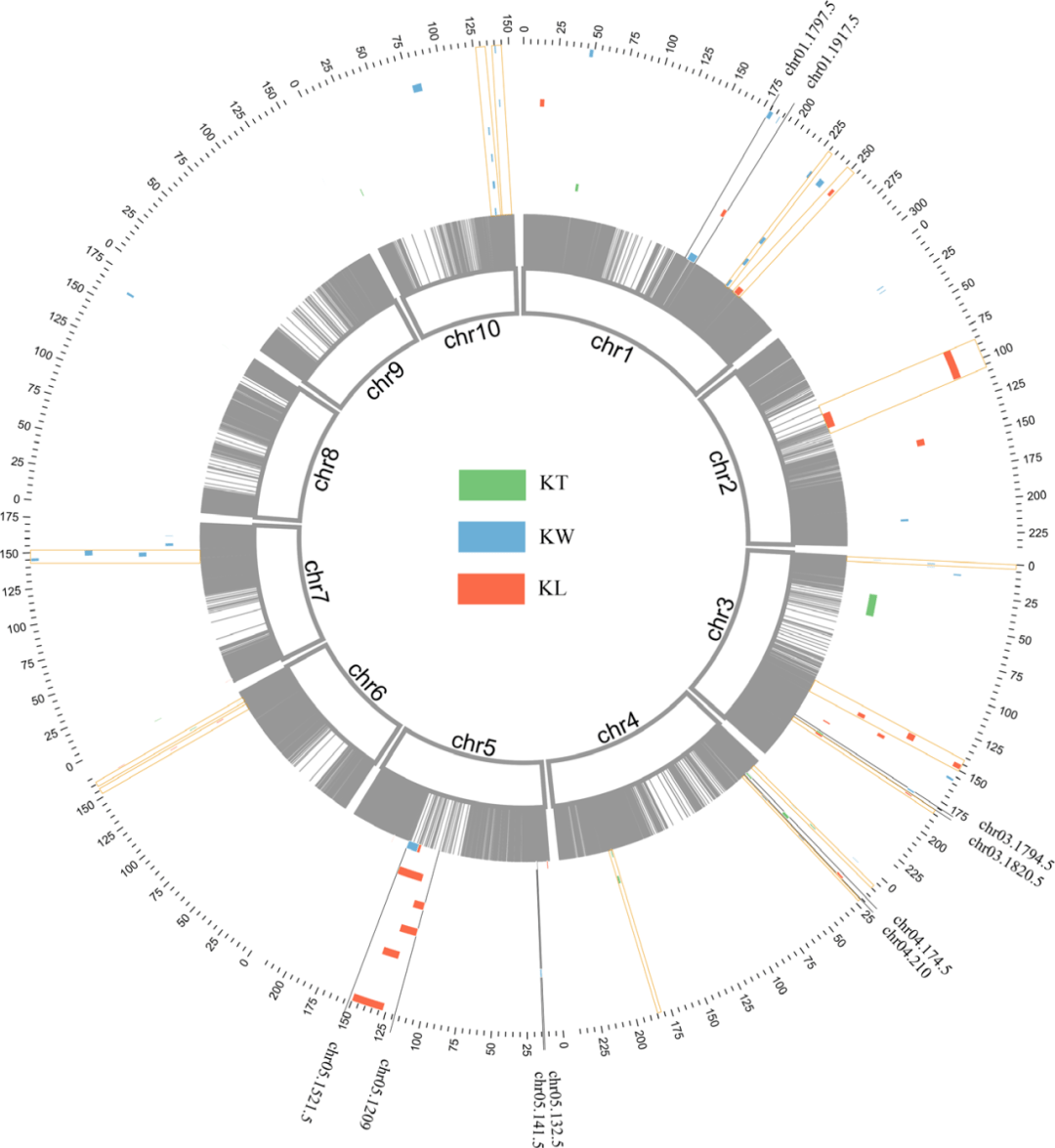
图3 QTL定位的结果
4. 好,第四点就是这篇故事的高潮了。由于本文的GWAS关联阈值降低一次之后,仍然获得了较少的显著关联SNP,为了能跟连锁分析的结果关联起来,作者又迫不得已降低了GWAS的阈值为1 X103 (肯定试了不同的阈值才选择了这个标准 )。基于SNP的物理位置信息,作者发现56个SNP能落入到18个QTL中,通过每个co-located上下游220 kb的区间筛选候选基因。通过同源比较后最终发现有50个和籽粒发育有关的候选基因,包括转录因子、酶或者转运蛋白。另外,作者发现一个有意思的现象是,这些区间中有7个miRNA,这些miRNA在拟南芥中的功能已经被研究清楚,参与叶片、花、种子,根系和叶绿素生物合成。Zma-miR399还跟玉米的低磷吸收有关。这个miRNA的筛选,可能是被逼出来的结果。这部分感觉是高潮,但是又有点意犹未尽。
5. 这部分也是这篇paper能发到PBJ的重要原因。有了这么多候选基因,总得找到一个靠谱的进行转基因验证(一区paper的落脚点)。为了能选择到一个靠谱的候选基因,作者基于已经发表的玉米籽粒发育转录组数据看候选基因的表达情况,令人难过的是,基于GWAS的17个候选基因在籽粒中均表现出差异表达;联合分析的候选基因中,35个候选基因中有29个差异表达。看样子,通过转录组缩小候选基因的方法走不通。得把目光放到其他的target上,由于miRNA只有7个,筛选起来也较为方便,继续利用已经发表的文章数据,发现两个miRNA表达量在籽粒中表达比其他5个高,而且,miR159还在拟南芥中调控胚乳的发育(好了,啥也不说了,下游验证就你了)。有了目标之后,RT-PCR,转基因就风风火火的开展起来了,具体的结果都是套路,笔者就不分享了。
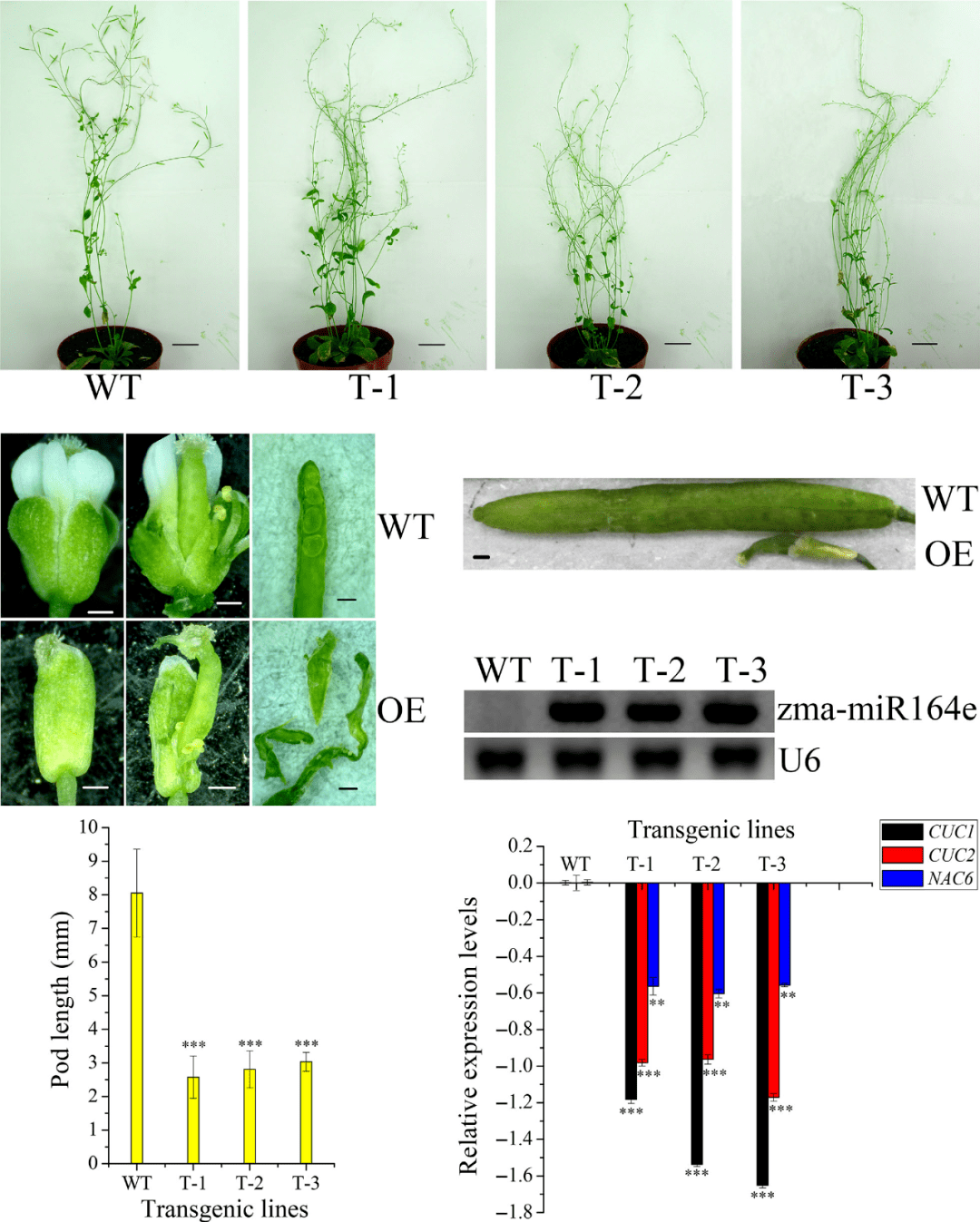
图4 功能验证结果
靠谱er的评价
槽点:
1. 表型鉴定中,使用了多种环境和重复,实际上还可以进行联合方差分析,看环境和重复的效应(这部分往往是个坑……有感兴趣的小伙伴儿想深入了解的请留言)。
2. 芯片获得的SNP密度不够,获得的显著关联SNP数量不多,导致确定显著关联SNP时,阈值一降再降。估计本文的作者心里也是多个MMMMP……在此,笔者真心不推荐GWAS采用芯片genotyping。
3. 候选区间确定的时候作者采用一刀切,上下游220kb的方法有待商榷,笔者认为,用LD block更严谨。
优点:
1. 表型数据鉴定工作量巨大,虽然本文只写了籽粒相关的性状,其他性状估计作者也考察了,弱弱的猜测下,后边该课题组肯定还有其他基于相同基因型,但是不同表型的结果发表出来。
2. 作者善于借力,本文的基因型(关联群体和连锁群体)均为已经发表的结果(自己课题组的数据),转录组的结果还是已经发表的结果,站在巨人的肩膀上,嗯,挺好。
3. 本文的表型数据和SNP关联/定位数据看上去较多,如果有类似实验设计的老师可以参考本文的写作方法,尤其是面对一堆数据如何从中筛选重要的结果进行描述以及结果描述的逻辑。
参考文献:
Zhang, X., Zhang, H., Li, L., Lan, H.,
Ren, Z., Liu, D., Wu, L. et al. (2016) Characterizing the population
structure and genetic diversity of maize breeding germplasm in Southwest
China using genome-wide SNP markers. BMC Genom. 17, 697.
转自:联川生物
- 本文固定链接: https://maimengkong.com/zixun/1472.html
- 转载请注明: : 萌小白 2023年4月24日 于 卖萌控的博客 发表
- 百度已收录
