当当当当,基迪奥有参转录组升级啦,升级的部分从最开始的比对参考基因组,到最实用的差异分析和富集分析都有。想要了解转录组分析点的朋友们,赶紧看过来吧~
首先,来看一下我们的分析流程,由于动植物的非模式生物与人大小鼠的模式生物的基因组、研究侧重点不一样,因此我们将报告拆分成了农学和医学的报告。
基于非模式生物的基因组不够完整的特点,我们会进行新基因鉴定和基因结构优化的分析;而由于人大小鼠的基因组非常的完善,这部分的分析可以直接省去,而融合基因与癌症息息相关,在人、大小鼠中研究的非常多,因此我们加入了这部分的分析内容。
此外,对于人、大小鼠,还有特定的数据库,因此也增加了对应数据库的富集分析,具体的分析流程参见下图1。
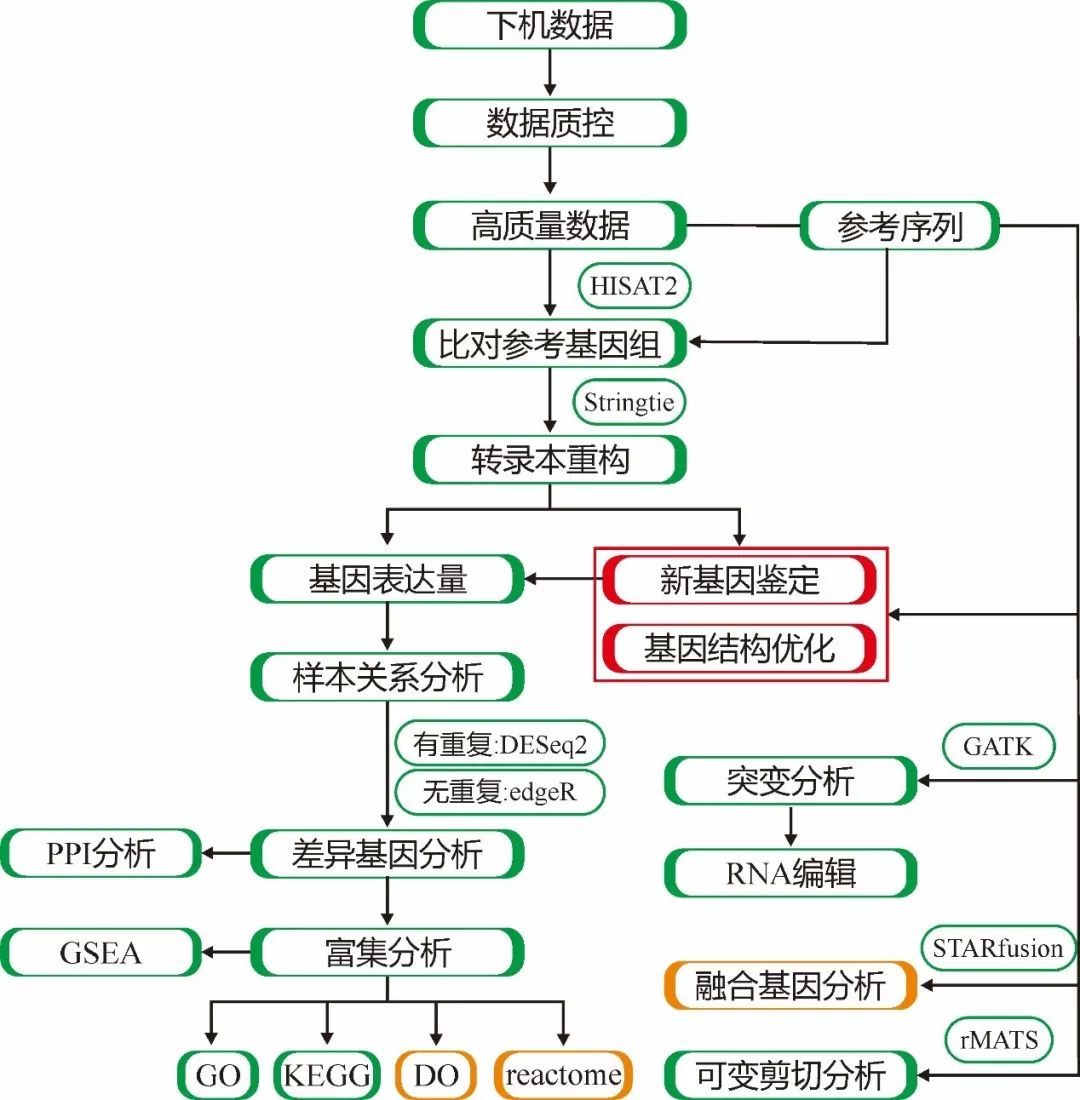
图1.有参转录组分析流程
其中,绿色的方框为所有物种共有的分析点,红色的方框是动植物(非人、大小鼠)所特有的分析点,而橙色的方框是人、大小鼠特有的分析点。
看到这么多分析内容,是不是想来了解一下呢,且听我们慢慢道来。
又快又准的流程分析
在一篇2017年nature communication的文章中[1],比较了目前转录组分析中39种分析软件,采用了120多种的组合,超过490个分析,最终给出了转录组分析流程中最合适的软件。我们根据这篇文章,对有参转录组的流程进行了优化。
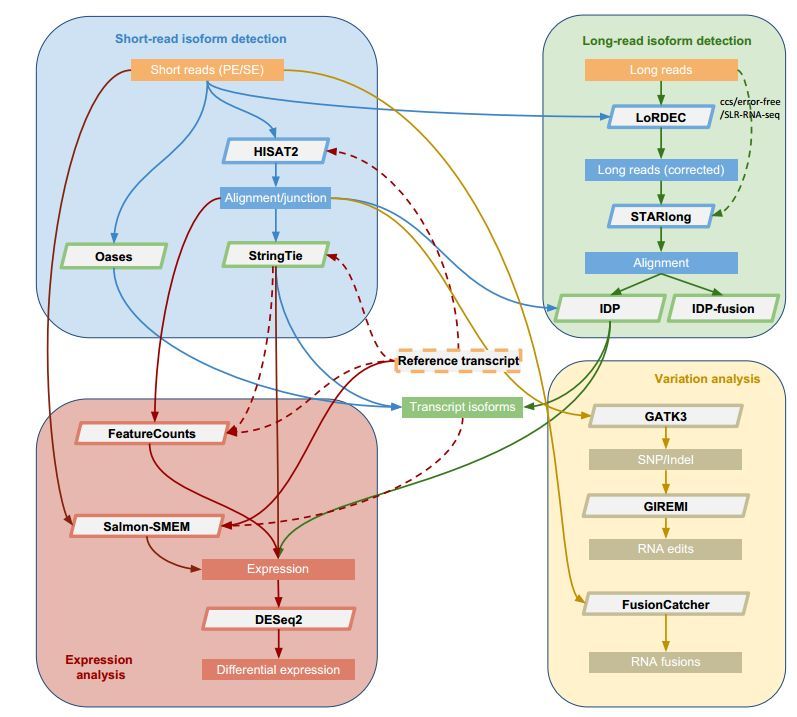
图2. NC文章建议的RNA-seq流程,蓝色背景是基于短reads的流程,即常见的有参和无参转录组,绿色的是基因长reads的流程,即目前的三代转录组,红色背景是表达量分析的部分,黄色背景的是变异分析的部分。
在做有参转录组的时候,比对参考基因组是分析的第一步,也是保证我们后续分析准确的最基本的一步,因此,选择合适的比对软件是非常重要的。
在NC的文章中,通过用不同的数据集对TopHat、STAR、HISAT2三个软件进行比较,最终得出结论,HISAT2是目前比对率最高且最准确的比对软件。除此之外,HISAT2还有一个非常厉害的优点,它的速度会比TopHat快上100倍。所以,更换了新的软件后,我们就可以更快的拿到更准确的结果了。
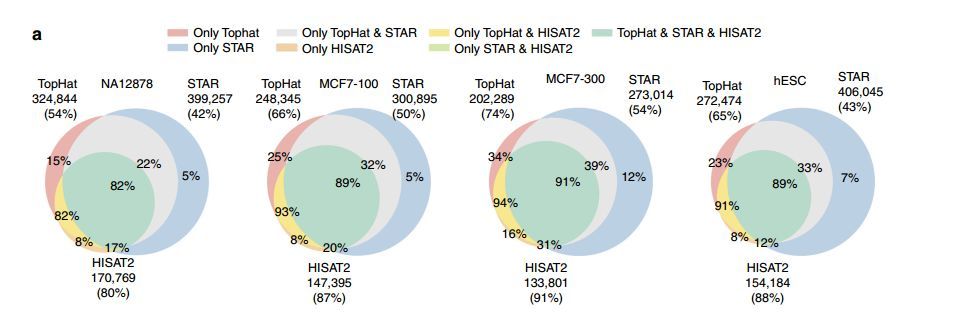
图3.不同样本用三个软件进行比对的比较
如果是非模式生物,由于参考基因组的不完整,我们还需要重构转录本,从而找出在测序中发现的新基因,并对已知的基因进行结构优化。转录本重构的常见软件有cufflink和Stringtie,NC的文章对此也进行了比较。
传统的cufflink在基于参考基因组的比对结果重构转录本时候,并没有考虑到转录本的丰度,容易造成转录本重构的错误以及数量的低估,从而影响结果的准确性。而Stringtie有效利用转录本的丰度信息,能够组装出更多的转录本,组装结果也更为准确。
因此我们选择了Stringtie作为转录本重构的软件进行分析。如果已经是人和大小鼠等模式生物,基因组已经研究的非常透彻了,基本没有什么新基因的存在,那么这一步就可以跳过,直接进行后续的分析了。
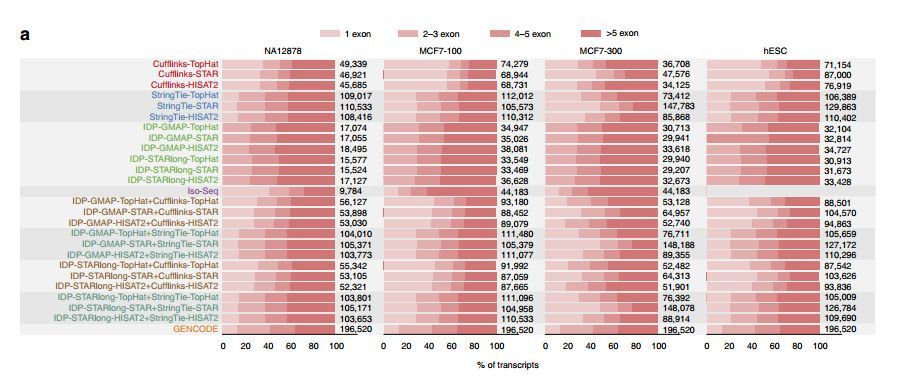
图4.采用不同转录本重构算法组合,转录本的外显子数分布
做转录组分析的重头戏之一就是差异分析了,由于各种客观情况的存在,会出现没有生物学重复的差异比较。
文章将DESeq2、limma、edgeR、cuffdiff等软件进行比较,研究发现,DSEeq2的性能是最好的,而在精确度上,DSEeq2和edgeR都有不错的表现。
由于DSEeq2没法针对两个样本间进行差异分析,而edgeR可以,因此,根据软件的特点,我们对不同情况选择不同的软件进行分析,在有生物学重复的时候,会选择用DESeq2进行差异分析;而在没有生物学重复的时候,就选择edgeR进行差异分析。
表2.差异分析软件
|
类型 |
软件 |
标准化方法 |
pvalue计算模型 |
FDR计算方法 |
差异基因筛选标准 |
|
有生物学重复 |
DESeq2(Anders et al, 2014) |
DESeq |
负二项分布 |
BH |
|log2(FC)| > 1,FDR< 0.05 |
|
无生物学重复 |
edgeR(Robinson et al, 2010) |
TMM |
负二项分布 |
BH |
|log2(FC)| > 1,FDR< 0.05 |
上面的内容都是软件的部分,可能大家还感受不出更新的特点,那么接下来说一下分析点增加的部分,这一部分可是非常重要的。
由于我们拿到手的结果是流程化的,并不能满足个性化数据挖掘的需求,经常还会提出补充分析。但从提出需求到获得结果都会花费时间,所以我们对补充分析进行了研究,将提出需求较多的分析点直接加入到了流程分析中,这样大大的减少了沟通和等待的时间。
基因互作关系寻找——我们有蛋白互作网络
由于基因与基因之间会有互作关系,而通常我们会利用到string数据库帮助我们进行研究,在以往的结果中,我们通常是挑好了目标基因后再去分析的,但有时候我们并不能找到目标,所以就会卡在这一步。
因此我们对全部差异基因进行了string互作网络分析,这样我们就能获得整体差异基因的互作情况,由于通常差异基因会比较多,所以做出来的网络图会密密麻麻的,这时候我们可以再进一步对这些结果进行筛选,这样就能快速的获得最终的互作网络图了。
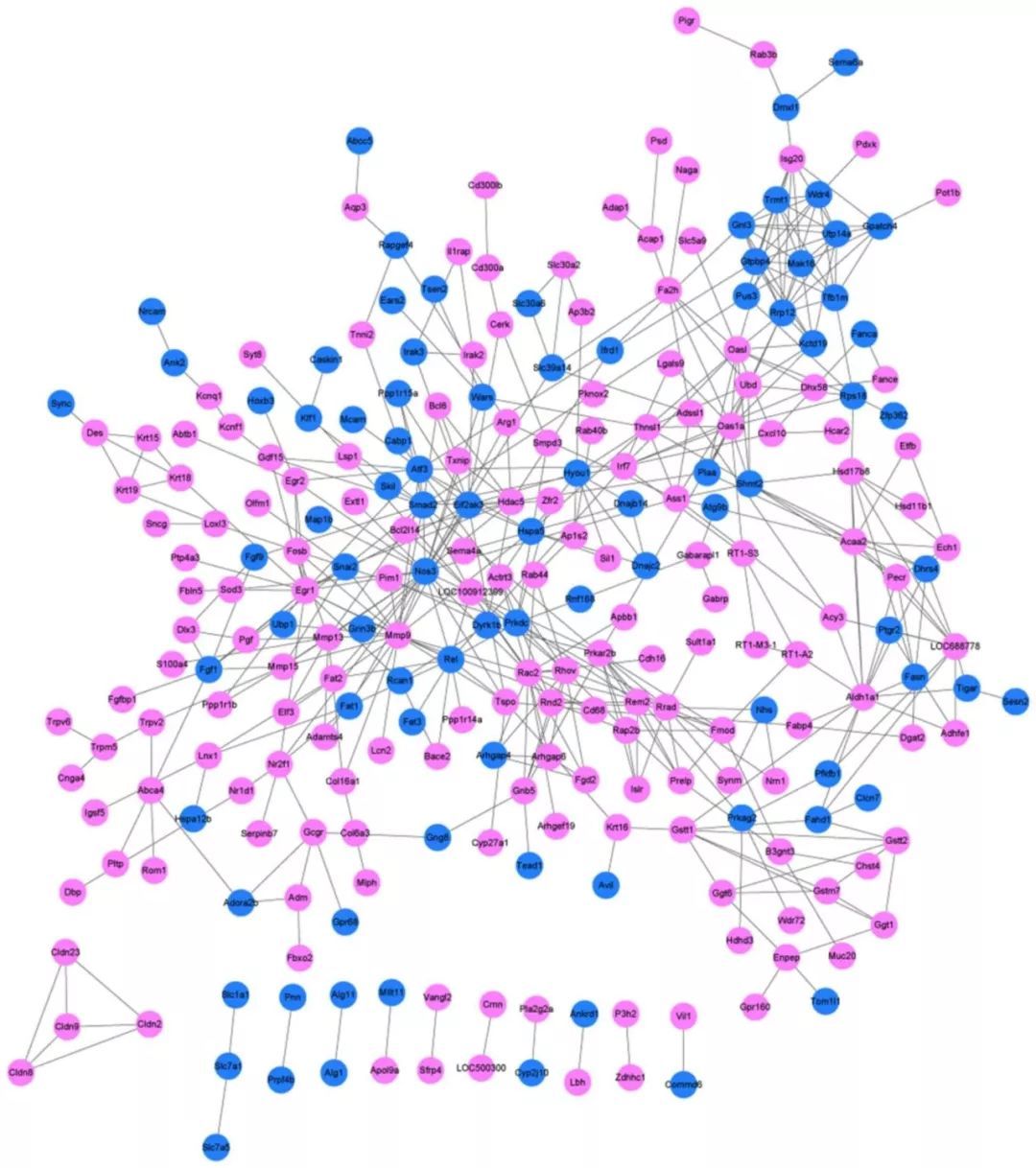
图5.网络图例图
与众不同的富集分析方法——GSEA分析在此
除此之外呢,功能分析一直是大家研究的重点所在,但是当我们的差异分析结果很少的时候,通过传统的超几何检验的富集分析得到结果可能会很少,甚至没有结果。
因此,为了能帮助大家更好的找到研究目标,我们特意增加了GESA的富集分析。GSAE的方法能够有效弥补传统富集分析对微效基因的有效信息挖据不足等问题,更为全面地对某一功能单位(通路、GO term或其他)的调节作用进行解释。
如果对GSEA的原理非常感兴趣的小伙伴们,可以听一下周老师的在线课堂哦(复制这里的链接到电脑端观看:http://www.omicshare.com/class/home/index/singlev?id=46)
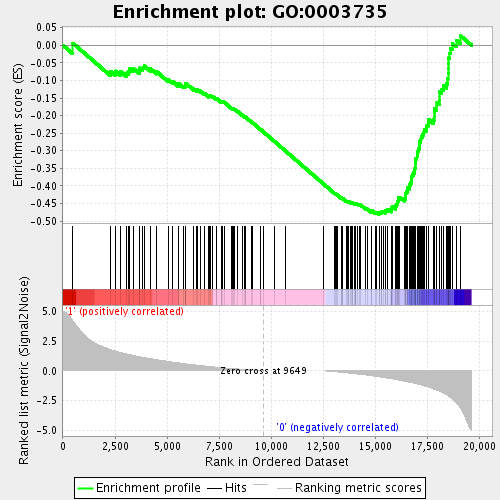
图6. GSEA富集结果
GO、KO还不够?我们有DO和Reactome相助
此外,在人、大小鼠的研究中,其实有非常非常多相关的数据库,根据我们的补充分析中数据库出现的次数比较,我们还额外增加了DO数据库和Reactome数据库的富集分析。
DO(Disease Ontology)数据库是描述基因功能与疾病相关的数据库。Reactome数据库汇集了人类各项反应及生物学通路,可以说是改良版的KEGG数据库。通过这两个数据库的富集分析,能更快的帮助我们找到与疾病或相关的差异基因。
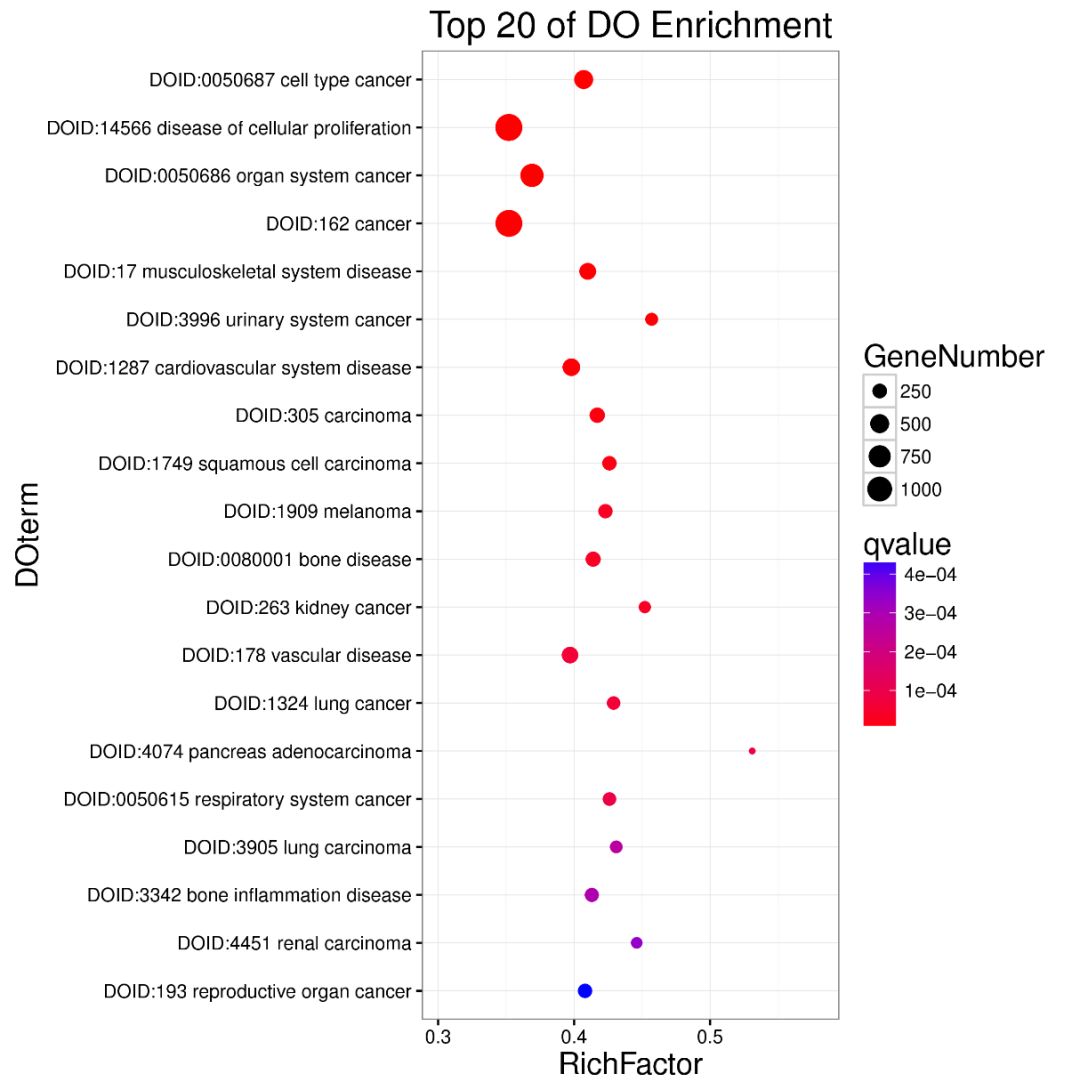
图7.DO富集分析
大家可以看到,我们此次报告的更新点是非常非常多的,并且十分贴合大家的需求,如果有意向做转录组的朋友们,赶紧联系我们吧~- 本文固定链接: https://maimengkong.com/moreshare/1114.html
- 转载请注明: : 萌小白 2022年7月9日 于 卖萌控的博客 发表
- 百度已收录
