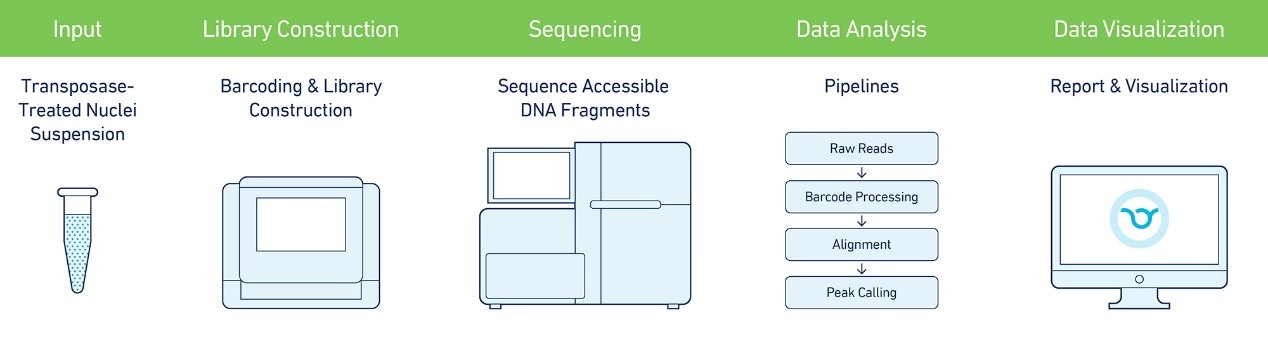
(图片来源:10x Genomics官方素材,侵删)
Loupe Cell Browser主要用于打开10X单细胞结果文件中的.cloupe文件,导入数据后,可捕获更多数据信息。在ATAC数据的应用主要在于:用于寻找显著peaks,区分转录因子motif,识别细胞类型,比较细胞群间染色质可接近性,以及探索细胞群内的亚群。
在开始学习教程之前,相信各位小伙伴们已经下载并安装了Loupe Cell Browser (https://support.10xgenomics.com/single-cell-atac/software/visualization/latest/what-is-loupe-cell-browser),和3’/5’基因表达谱数据可视化一样,同样使用的Loupe Cell Browser 3.3.1。
大家可以用10x官方ATAC教程数据集或自己的10x scATAC数据跟着我们一起测试。
10x官方ATAC教程数据集
打开界面后你可以通过点击“Recent Files”页面中的“ATACTutorial.cloupe”来获取ATAC教程数据集。ATAC教程数据集是根据标准Chromium™ Single-Cell ATAC protocol利用cellranger-atac流程分析人外周血单核细胞得到的结果,流程分析得到的barcode计数为5335。
自己的10x scATAC数据
点击下方Browse for a Loupe Cell Browser File按钮打开新的cloupe文件。在我们公司所提供的10×ATAC的分析结果中,每一个样本都有对应的.cloupe文件,Loupe Cell Browser安装后可以直接点击sample.cloupe文件打开并查看相应结果。

打开后我们可以看到多种操作工具,运用这些工具我们可以实现五大应用,本次Dr.cell先带各位先熟悉使用loupe软件鉴定细胞类型和分析差异可接近性两大应用。:
1. Identifying Cell Types 鉴定细胞类型
2. Analyzing Differential Accessibility 分析差异可接近性
3. Exploring Cell Subtypes 探索细胞亚型
4. Finding Significant Features 发现显著性特征
5. Sharing Results 分享结果(输出想要的图表)

相信各位已经使用过Loupe Cell Browser来分析10×单细胞基因表达,当各位开始着手分析10×ATAC数据时,会发现与10×单细胞基因表达既有相同之处,又有不同之处。
Cell Ranger ATAC算法文档包含了更多算法和分析方面的细节,敲黑板,以下是在loupe上查看10×ATAC数据时值得注意的关键性要点:
① UMI count per cell是基因表达的单位。Cut sites per cell是染色质可接近性的单位。
② 基因表达矩阵中每一行是基因。染色质可接近性矩阵中每一行是peak。
③ Peaks是基因组区域,这些区域在片段切割位点(fragment cut sites)显著上升,即表明为开放染色质区域。它们通过其位置来命名(例如“Chr1:10244-10510”)。
④ 通常,ATAC数据集中的peaks数要比参考基因组中的基因多。
⑤ 除了peaks之外,还有一些其他累加特性类型可用来进行细胞区分:
启动子总和(Promoter sums),是接近该基因的转录起始位点之一的cut sites per cell (within peaks)的总和。这些特征被命名为"(Gene)Sum"。并非所有peaks都被关联到了一个基因。
转录因子motif(Tranion factor motifs),是位于被Cell Ranger ATAC流程关联了motif的peak中的cut sites per cell的总和。Motif特征是以motif本身命名的(例如“SPI1”)。一个peak通常会关联多个motifs。
1 Identifying Cell Types 鉴定细胞类型
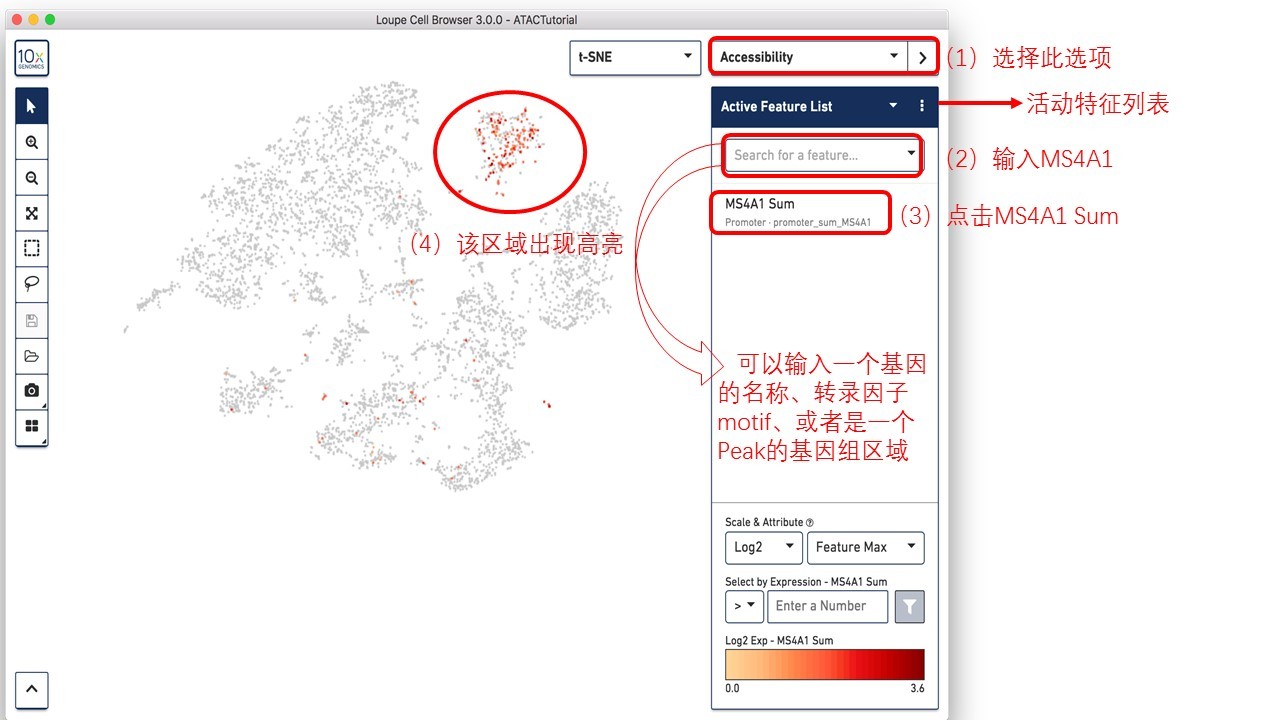
根据已知的markers识别细胞类型非常直接,快速。可以利用启动子总和确定细胞类型,首先,在模式选择器中选择Accessibility Mode(可接近性模式)进行操作,可以看到一个Active Feature List(活动特征列表)。
例:就B细胞的marker MS4A1而言,在搜索框中输入“MS4A1”,检索得到“MS4A1 Sum”特征。按下Tab键或回车键来将启动子总和加入活动特征列表,并计算整个数据集中该启动子的切割位点数。我们发现B细胞marker明显地聚集在图中一群中,并呈高亮模式。
接下来,可以通过添加B细胞的其他markers,如CD19和IGKC,将其启动子总和加入到活动特征列表,可确定高亮的区域就代表B细胞。
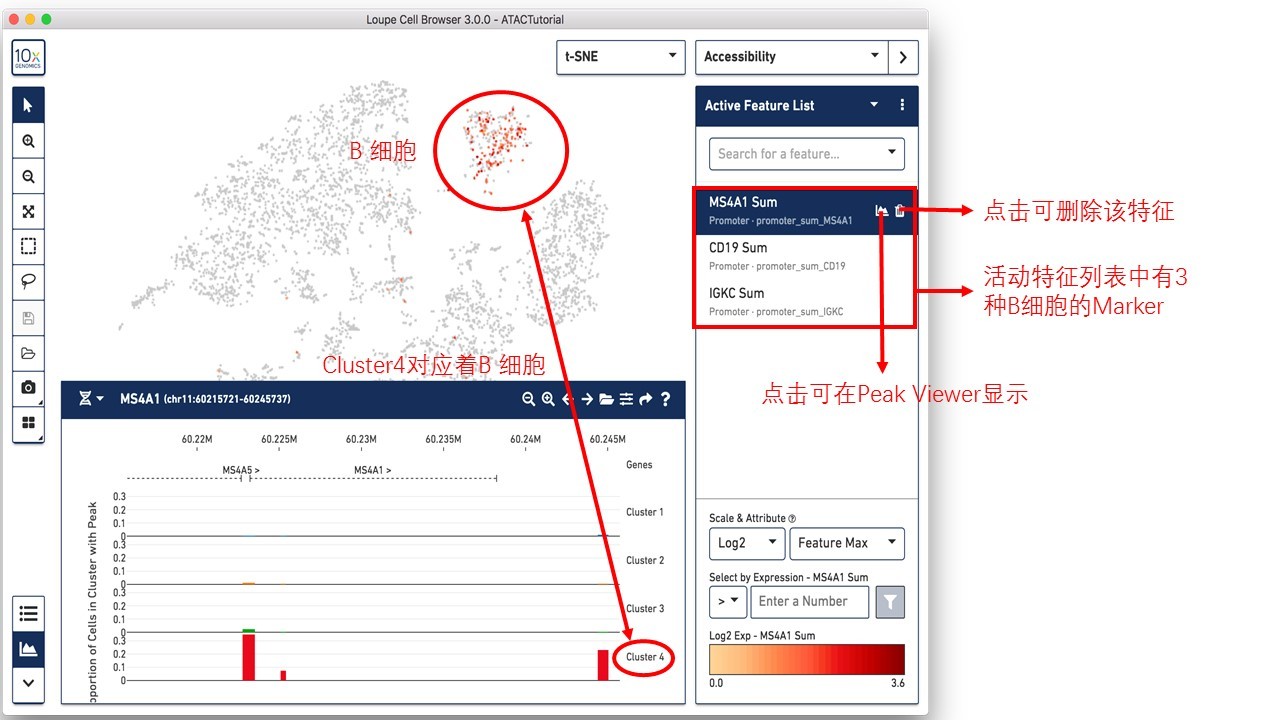
除了利用启动子总和来确定细胞类型,还可以利用转录因子motifs确定细胞类型。研究表明,SPI1(PU.1)转录因子在单核细胞功能中扮演了关键角色[1], 选择将SPI1加入到活动特征列表,将高z-score的细胞显示为红色,表明在所有具有SPI1 motif的peak之间具有较高的相对可接近性。SPI1在B细胞调节中也具有一定作用,由于我们已经通过B细胞的marker(MS4A1、CD19和IGKC)标记识别了B细胞分群,因此,左上方的大群应该是单核细胞。

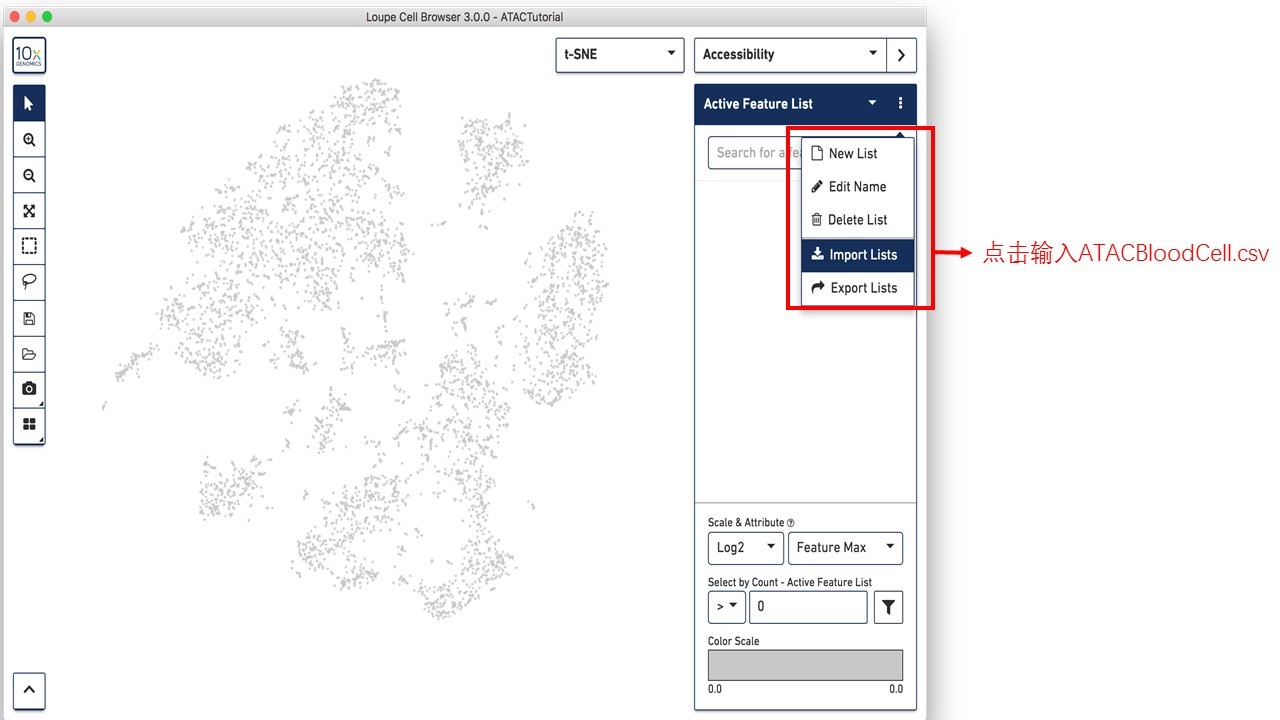
和利用loupe软件查看10×单细胞基因表达数据类似,除了直接在搜索框中输入基因symbol外,还可以导入感兴趣细胞类型marker的CSV文件(ATACBloodCell.csv),CSV文件内容如下截图所示。

接下来,我们可以尝试创建一个B细胞分群。在工具箱中选择矩形套索工具,拖动选框选中我们之前通过输入B细胞marker(MS4A1、CD19和IGKC)高亮的细胞群,会弹出一个对话框,如下图填写名称,将这些细胞命名为“B细胞”。按下保存按钮,一个新的细胞类型分类就创建成功了。可以采取同样的方法创建一个新的“单核细胞”分群。
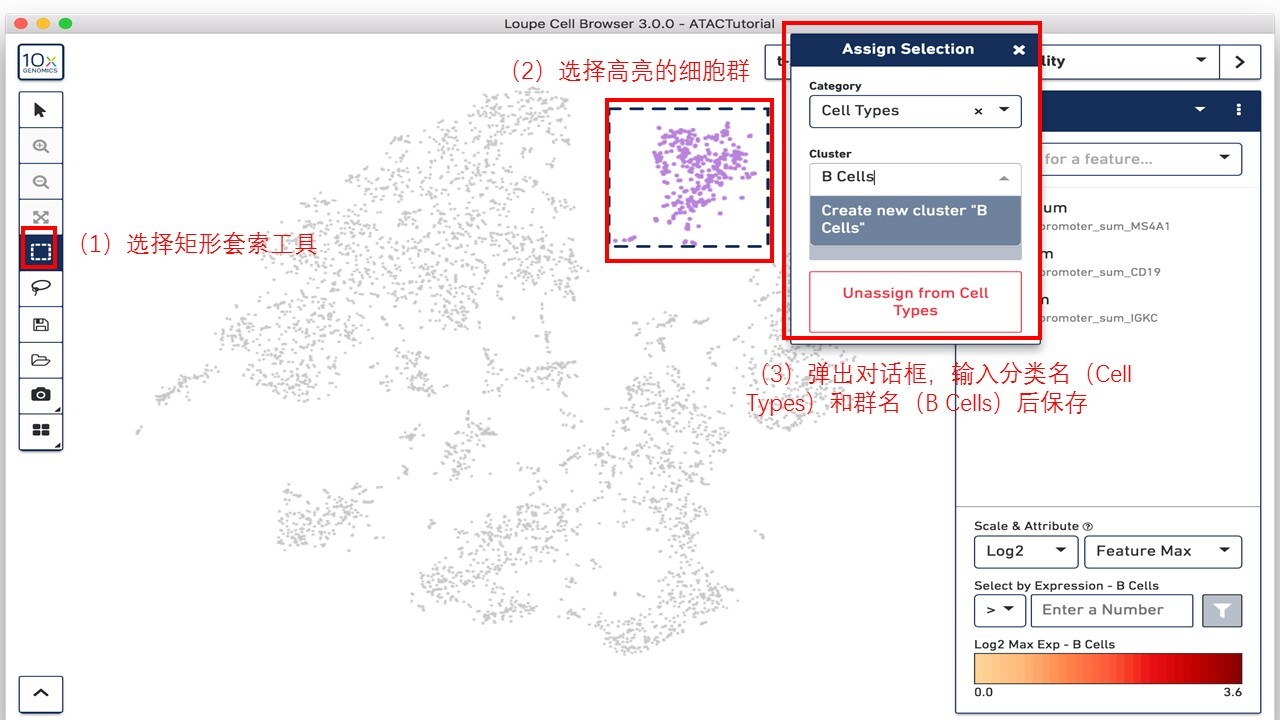
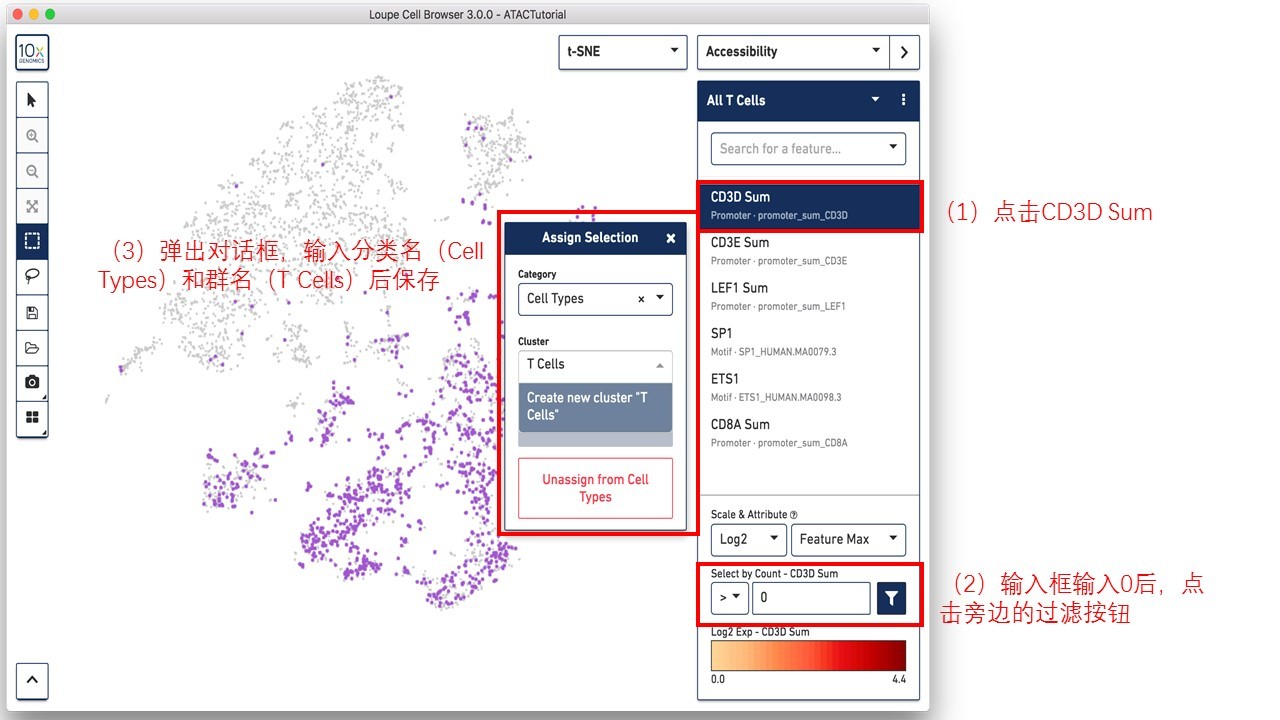
也可以定量的创建细胞分群,选择从 ATACBloodCell.csv 中导入的All T Cells,点击列表中的CD3D Sum,随后在“Select by Count - CD3D Sum”下方的输入框输入“0”,点击过滤按钮。这将会高亮含有CD3D启动子peak内存在fragment的每一个细胞,并弹出一个分群的对话框。选择“Cell Types”作为分类,将这些细胞加入到“T Cells”分群中。如下图所示。
2 Analyzing Differential Accessibility 分析差异可接近性
在1中创建了以 “Cell Types”为分类,共3种细胞群,即B细胞、T细胞和单核细胞。可以利用Peak Viewer分析差异可接近性,在选择器中选择Gene(基因)选项,输入 “CD33”,得到下图结果。

用鼠标点击peak,可以看到关于该peak细胞类型的百分及其他重要信息。单击一个peak也会在barcode图中展示在该区域拥有开放染色质的细胞,如下图所示。很明显,相较于其他细胞类型,很高比例的单核细胞在该peak区有开放染色质。

在我们公司Cell Ranger ATAC流程生成的fragments.tsv.gz文件可以在更精细的分辨率下查看可接近性,主要查看切割位点信息。通过文件夹图标可加载报告中生成的fragments.tsv.gz文件,Fragment文件可以通过文件系统或URL加载。这里,可以加载10×官网提供的ATACTutorial fragments文件,结果如下图。

3 Exploring Cell Subtypes 探索细胞亚型
本应用的目标:探索数据集中已知的未成熟(immature)和成熟的(mature)的B细胞类型。
上篇我们已经使用MS4A1和CD19的启动子总和鉴定了B细胞分群,通过在已经鉴定B细胞分群的基础上创建未成熟和成熟B细胞的亚型。小伙伴们好奇怎样去创建未成熟和成熟B细胞的亚型吗?请听Dr.cell娓娓道来。
小知识:什么是TCL1A基因?
它是在未成熟B细胞中表达而在成熟B细胞中不表达的基因,所以可以利用TCL1A 启动子总和来区分B细胞亚型
▶ 构建Filter
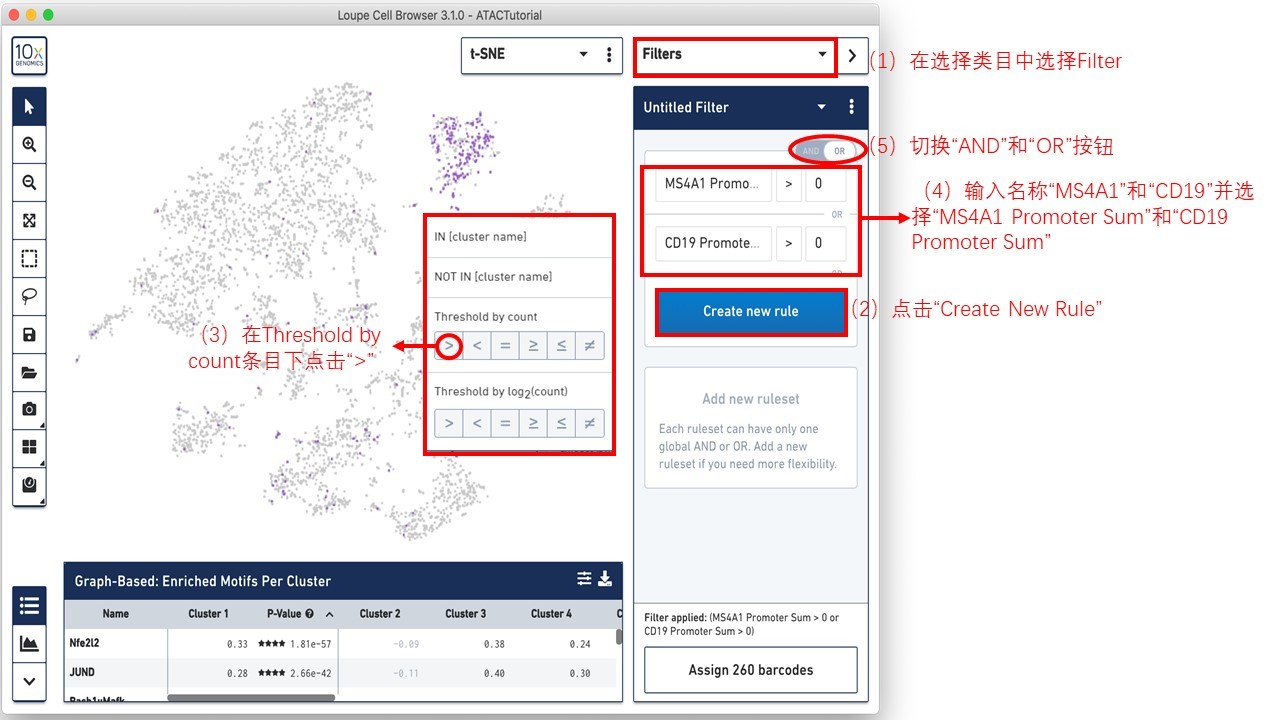
首先,在选择类目中选择Filter,随后开始构建一个Filter。通过点击loupe软件中“Create New Rule”图标增加过滤规则。因为我们想要设置启动子总和>0的阈值,所以在Threshold by count处点击“>”按钮,后输入名称“MS4A1”并选择“MS4A1 Promoter Sum”。CD19也是同样的操作。当尝试将“AND”按钮切换到“OR”按钮,立即会看到B细胞分群中更多的barcodes以紫色高亮显示(如下图所示)。
▶ 寻找未成熟的B细胞
我们希望找到一群MS4A1或CD 19启动子总和大于0并且表达TCL1A基因的barcodes。首先点击“Add new ruleset”按钮,接着点击“Create new rule”按钮,在Threshold by count处点击“>”按钮,后输入名称“TCL1A”并选择“MS4A1 Promoter Sum”,从barcode图中可以看到未成熟的B细胞呈紫色高亮。

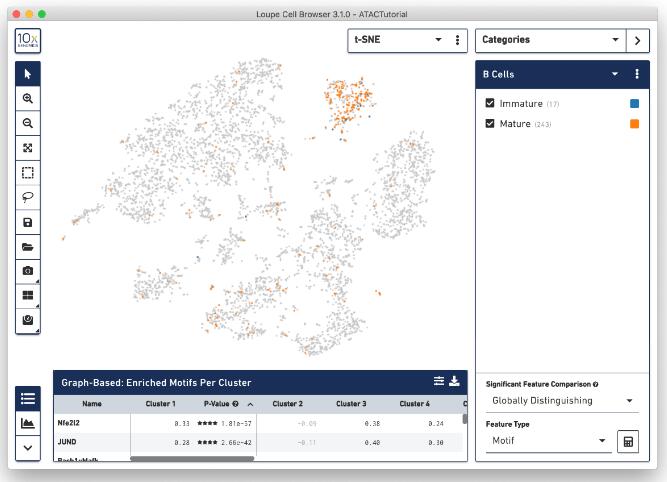
如下图所示,通过点击"Assign 17 barcodes"按钮,将这些细胞分配到一个category,命名category为 "B Cell Subtypes",命名cluster为"Immature"。由于成熟B细胞中不表达TCL1A基因,可以将"TCL1A Promoter Sum >0"修改为"TCL1A Promoter Sum =0"。设置完毕后可以在barcodes图中看出B细胞分群中有部分细胞呈紫色高亮。接下来通过点击"Assign 243 barcodes"按钮,将这些细胞分配到命名为"B Cell Subtypes"的category中,并将cluster命名为"Mature"。

最后,未成熟和成熟B细胞的亚型如下图所示。这样,我们就成功地使用loupe软件探索了细胞亚型。
4 Finding Significant Features 发现显著性特征
我们可以看看barcode图,看看带有T细胞标记的细胞区域。通过LSA降维和随后的聚类将T细胞分成几个单独的群组,如t-SNE图所示。是什么导致了这些亚群之间的差异?我们可以使用显著性特征工具来找出答案。
首先,利用套索工具高亮T细胞最右侧(rightmost)的分群并将其标记为“T细胞2”,如下图所示。

loupe软件显著性特征工具?
使用此工具,我们可以在当前选定的Clusters之间计算区别motifs,独立Peaks,或启动子总和。
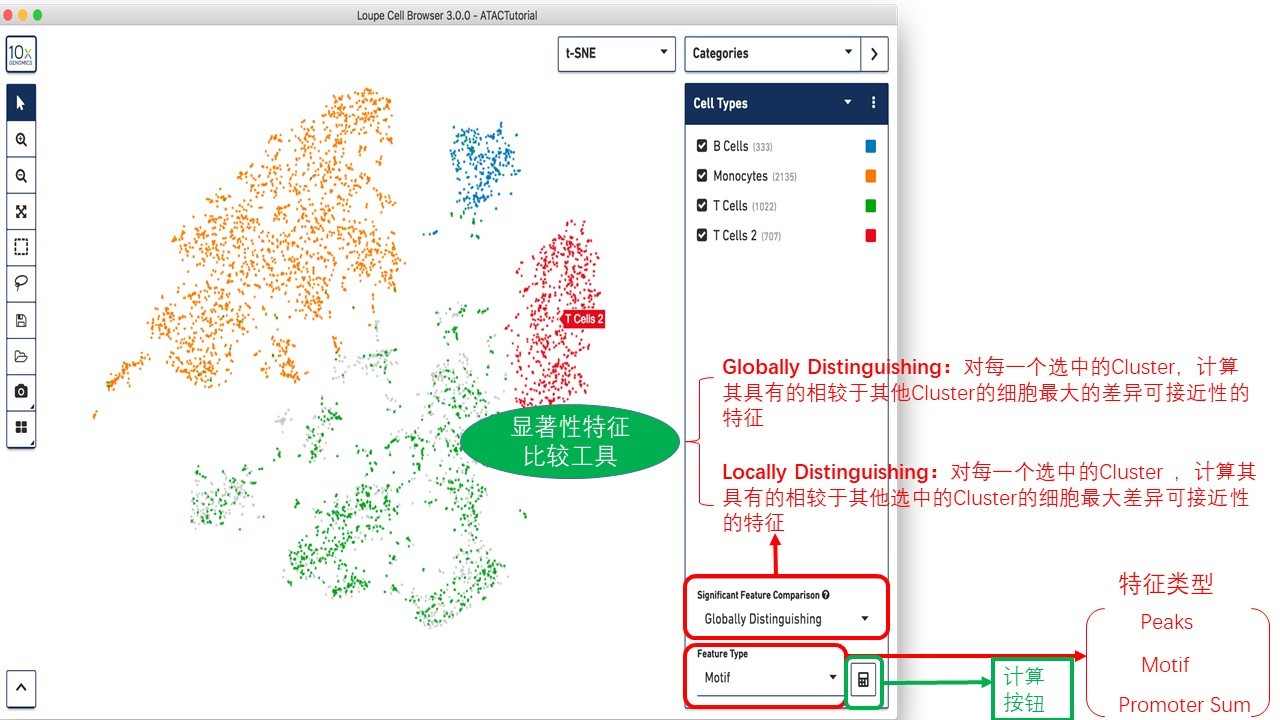
▶ 使用显著性特征工具来计算每个细胞类型的区分启动子总和
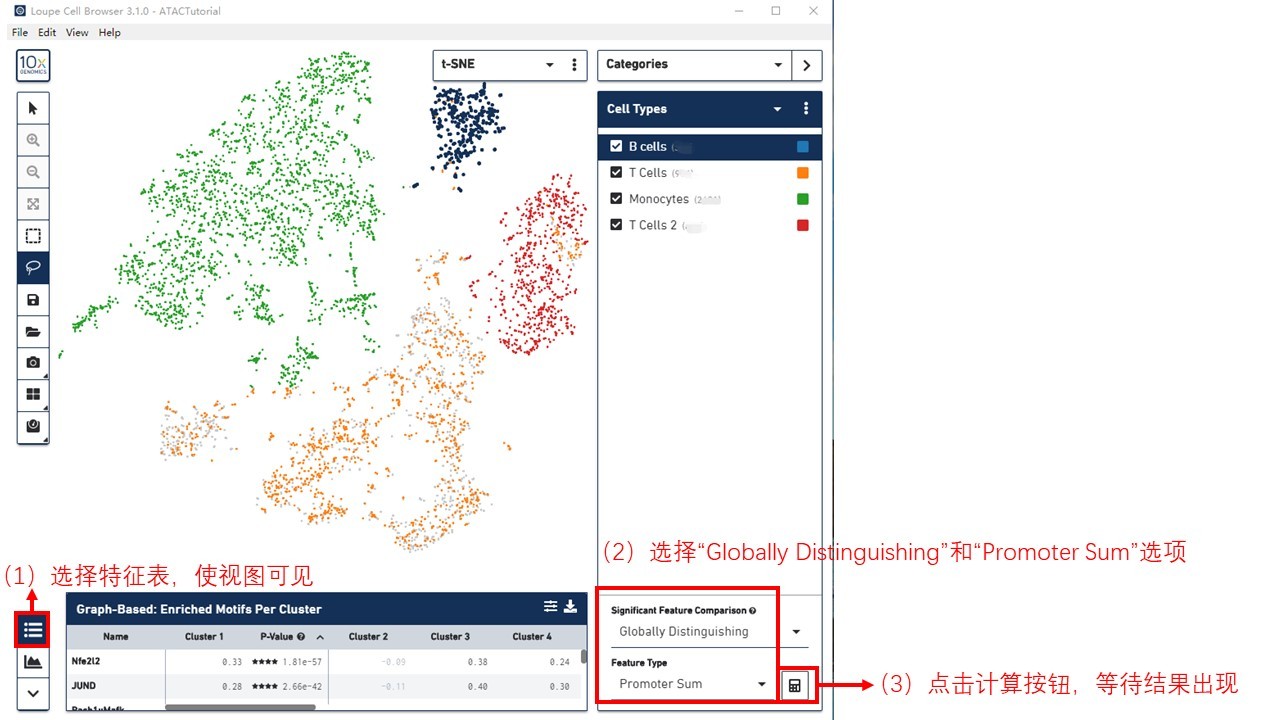
首先,单击底部面板左侧的列表图标,使特征表格视图可见。
接着,当处于分类模式下,Cell Types可见时,在显著性特征比较工具中选择Globally Distinguishing,选择启动子总和(Promoter Sum)作为特征类型。点击计算按钮,等待 Loupe Cell Browser计算显著富集启动子总和。
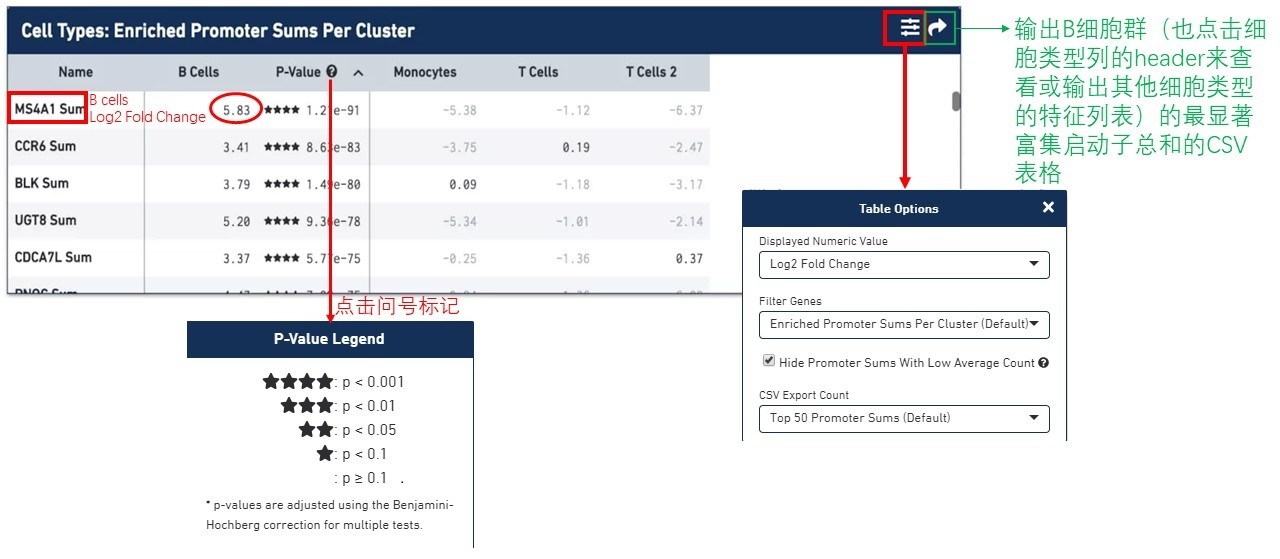
当计算完成后,出现B细胞群的最显著富集启动子总和,以及根据p-value排序的,且相较于其他细胞群的log2 fold change值。毫无意外,最显著富集的启动子总和就是我们用来鉴定B细胞分群的MS4A1(CD20)。如下图所示。
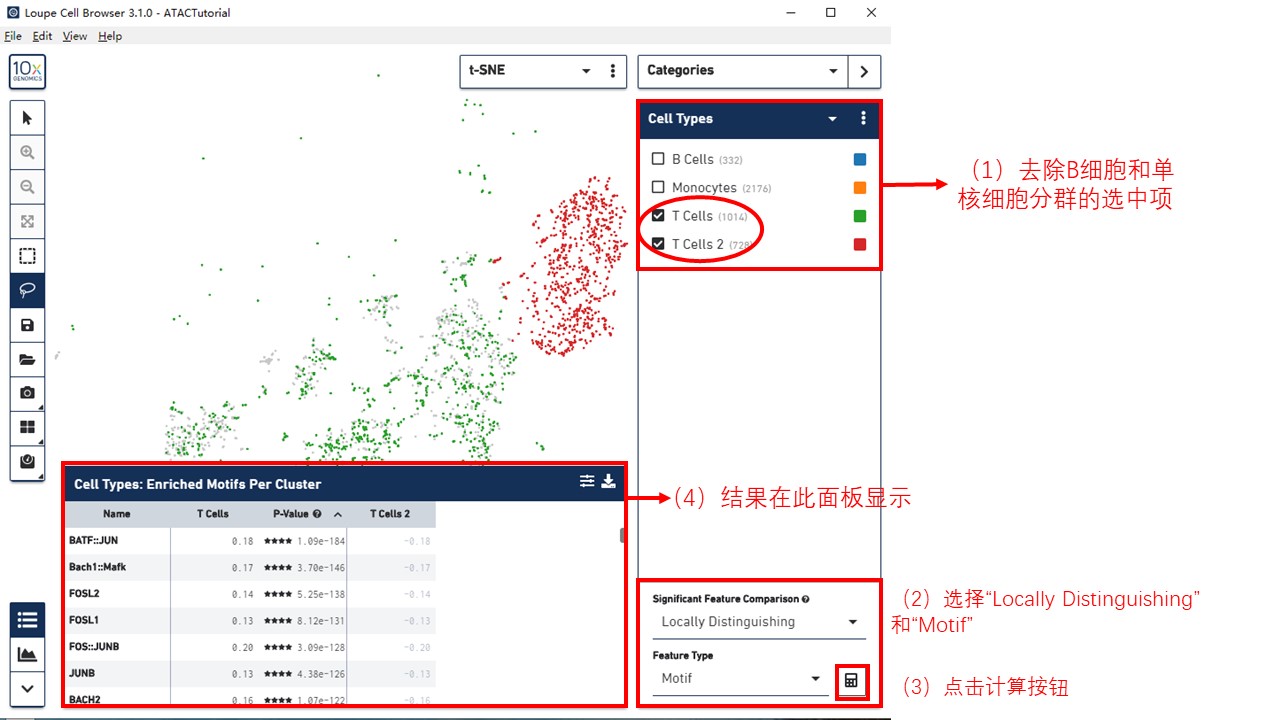
▶ 两个T细胞分群(T Cells and T Cells 2)之间的差异
在Cell Types面板中,去除B细胞和单核细胞分群的选中项,接着在显著性特征比较工具中选择Locally Distinguishing,将Motif作为特征类型,点击计算显著Motif。得出结果如下图所示。
在工具箱底部点击分屏按钮,选择Other Category(Cell Types),barcode图中的细胞将会以Cell Types(细胞类型)进行分屏显示。
接着,在特征表格中点击T Cells中最显著富集的motif “BATF::JUN”,将其加入活动特征列表,可在barcode图中查看BATF::JUN motif的z-score值。

从图中可以看出,T Cells 2组中的细胞在颜色上相对更蓝一些,表明该组中BATF相关peaks相较于T Cells组,其平均可接近性更低。研究表明,BATF motif可接近性在细胞分化和衰老中表现出增加趋势[1],因此这也可能说明T Cells 2组中的细胞相对更年轻,或者说包含更多原态T细胞(naive T cells)。
▶ 两组T细胞之间(T Cells和T Cells 2)的显著性peaks
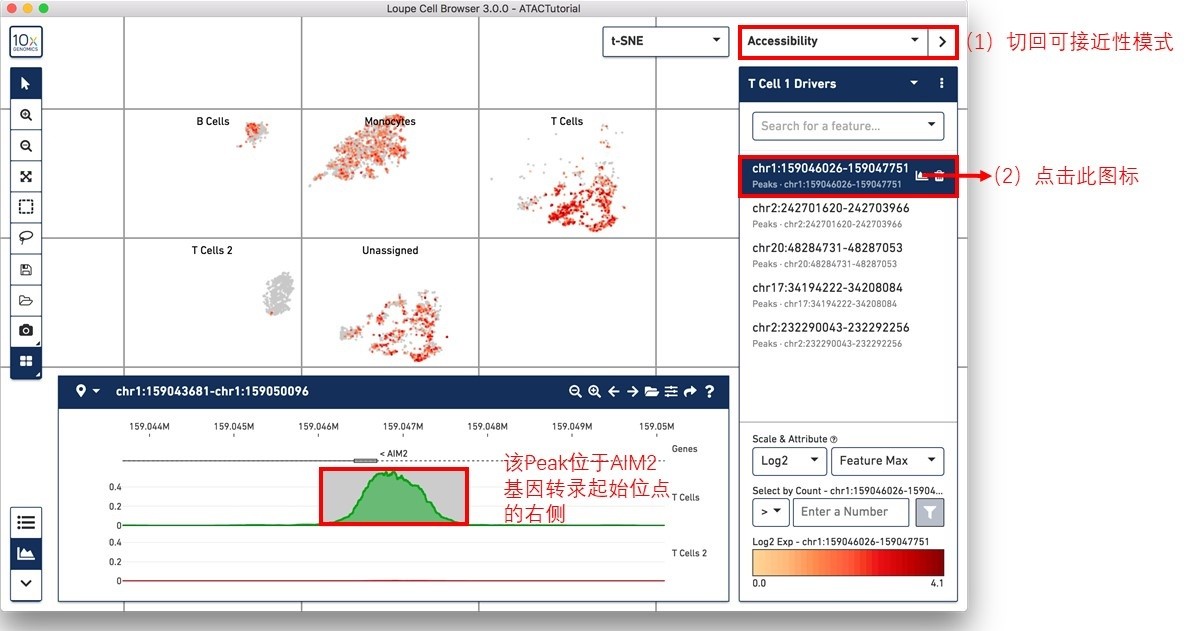
切换回分类模式,选择Peaks作为特征类型。点击计算按钮,等待计算完成。随后点击显著富集的前5个Peaks(如下图),将其逐次添加到新的“T Cell 1 Drivers”列表。关于新的“T Cell 1 Drivers”列表的创建,即在Add to Feature List输入区输入“T Cell 1 Drivers”即可。

为了了解这些独立的Peaks,我们可以将分类模式切换成可接近性模式,选择列表中的第一个Peak,“chr1:159046026-159047751”,点击Peak Viewer图标,如下图展示。在Peak Viewer中点击放大或缩小按钮可以显示其他附加信息,从基因注释轨迹中看到该Peak位于AIM2基因转录起始位点的右侧。
5 Sharing Results 分享结果
上述各种神操作,各位小伙伴们一定好奇怎样将结果保存下来呢,请继续跟随Dr.cell的步伐。从我们10×ATAC数据集中导出数据和图表有多种方法。工具箱、分类列表、特征列表、特征表以及Peak Viewer都具有导出功能。下面来一一阐述。
▶ 导出Barcode图
导出前的设置如下图所示。点击工具箱中的导出屏幕图形图标将会导出现在展示的barcode图。你可以选择导出为PNG格式或SVG矢量图格式。

▶ 导出特征表(Significant Features)
导出前的设置如下图所示。导出包含p-values 值和 log2fold changes值的CSV文件。

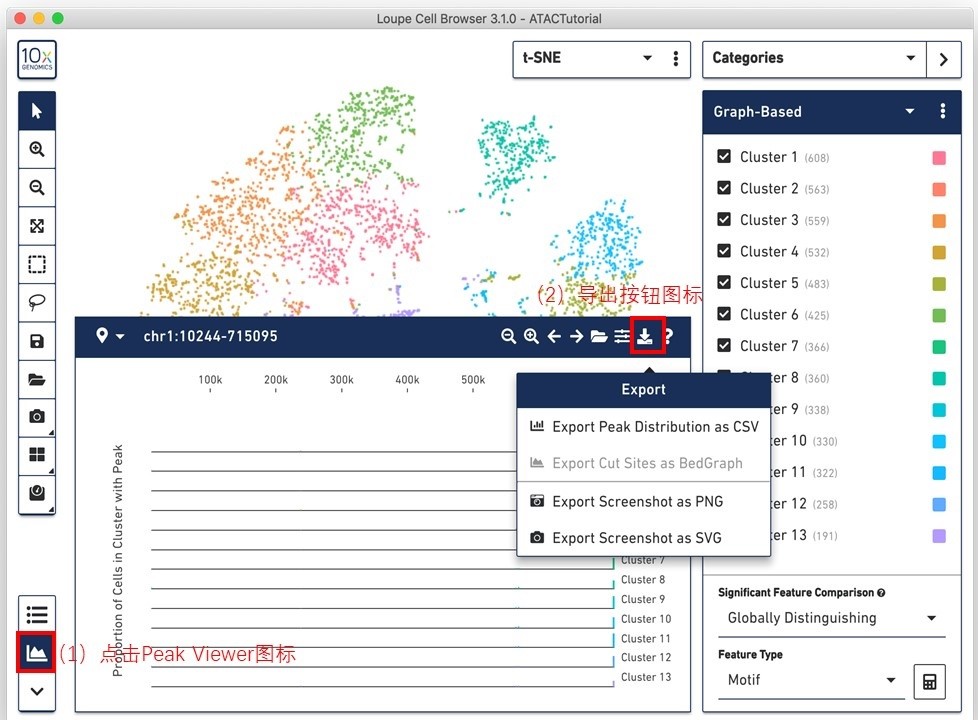
▶ 导出Peak Viewer图
导出前的设置如下图所示。可导出当前可视化Peaks到一个CSV文件中,或者将Peak Viewer图导出为PNG或SVG格式。
参考文献:
[1] Takahiro S , Mika N I , Haruka Y O , et al. Reconstruction of Monocyte Tranional Regulatory Network Accompanies Monocytic Functions in Human Fibroblasts[J]. PLoS ONE, 2012, 7(3):e33474.
[2] Moskowitz D M , Zhang D W , Hu B , et al. Epigenomics of human CD8 T cell differentiation and aging[J]. Science Immunology, 2017, 2(8):eaag0192.
- 本文固定链接: https://maimengkong.com/learn/1512.html
- 转载请注明: : 萌小白 2023年5月3日 于 卖萌控的博客 发表
- 百度已收录
