大家好,我是Leopard。
大家在处理生物信息数据,比如RNA-seq数据、芯片数据,或者从TCGA数据库下载的数据时,都会遇到各种各样的ID!这些ID就像是我们每个人的身份证,用于对基因进行标识。由于ID来自于不同的数据库,或者说命名的意图不同,所以对于同一个基因,总是有多个不同的ID,最常见的比如entrze ID、ensembl ID、HGNC ID、refseq ID等。
Entrze ID是美国NCBI数据库中的基因标识符,通常是由纯数字表示,比如人类TP53基因的Entrze ID是7157(注意,不同物种的基因ID是不同的);ensembl ID是欧洲生物信息数据库的基因标识符,都是以ENSG(ensembl gene)四个大写字母开始,后面跟着11位数字,所以ensembl ID的长度通常都是15位,比如人类TP53基因的ensembl ID是ENSG00000141510,值得注意的是ensembl ID不仅包含了两万多个蛋白质编码基因,同样也有很多的假基因、miRNA等,因此它的数量较多,有六万多个,比人类已知的基因数多得多;HGNC ID 是指由人类基因命名委员会(HUGO Gene Nomenclature Committee)指定的基因标识符,该委员会通常对基因赋予一个名字以及一个ID,比如人类TP53基因,其标准的symbol是TP53(相当于简称),标准的名称是tumor protein p53,HGNC ID 是11998。Refseq是美国NCBI提供的基因标准序列(参考序列)数据库,在该数据库中,人类TP53基因的ID是NG_017013。
此外,与某个基因相关的还有GO ID、芯片探针ID、uniprot ID等。所以,当面对众多的ID时,很多初学者是倍感头疼的,明明刚才还很熟悉,穿上个马甲就不认识了。

而且当我们在发表文章时,在文章中呈现的通常都是基因的HGNC symbol,就像TP53,EGFR,KRAS。所以,在进行数据分析前,熟悉ID转换至关重要!
本期,Leopard将给大家介绍Biomart数据库以及R语言中配套的biomaRt包的使用方法,帮助大家轻松实现ID转换。
Biomart是ensembl下属的一个网络数据库,里面包含非常多的信息。biomaRt包是该网站的R语言接口,可以帮助用户在R语言中实现biomart的功能,因此使用该包时必须保证互联网连接通畅!下面将给各位看官介绍biomaRt包的使用方法。
一、包的下载与安装
与其他所有bioconductor包一样,biomaRt包的下载也遵循同样的方法:

二、选择目标数据库(mart)和数据集(dataset)
Biomart目前提供了四种数据库,可以使用listMarts()函数查看:
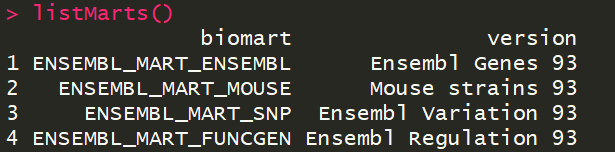
当处理人类基因时,我们利用useMart()函数选择第一个“ENSEMBL_MART_ENSEMBL”,并将其赋值给,my_mart对象。代码如下:
注意,此处引号中写全称“ENSEMBL_MART_ENSEMBL”也可以!
在ensembl数据库中包含了77个数据集,大家可以使用以下代码查看:
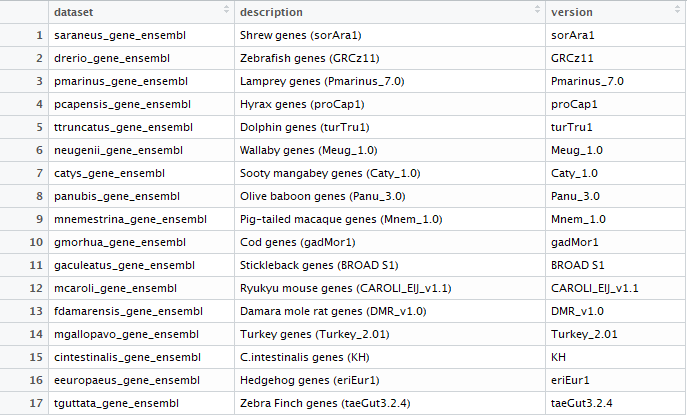
为了进一步操作,我们必须从这77个数据集中选择一个数据集,此处当然是选择人类基因的ensembl数据集咯~代码如下:
至此,数据集的工作已经完成了,下面我们就可以在此数据集中,根据需要进行ID的转换。此工作由getBM()函数完成。该函数有四个参数,通常情况下都需要指定!
1)attributes参数:该参数可以接受一个字符串向量,用来指定输出的数据类型,就是你要什么,比如entrezgene,hgnc_id。
2)filters参数:该参数也可以接受一个字符串向量,用来指定数据的输入类型,比如你的原始信息是基因的ensembl ID,并且有这些基因的染色体位置信息,那么此处的filter就是ensembl ID和chromosome_name等。
3)values参数:与filters参数一一对应,即设定filters的具体的值,也就是你的原始数据。
4)mart参数:此前定义的数据库,此处就是my_dataset。
attributes和filters参数具体的取值可以通过listAttributes()和listFilters()函数查看。只需要在函数中定义mart参数为我们当前选择的my_dataset即可。
下面我们进行几个实战演练。
三、根据ensembl ID获取基因名
假设我们有六个基于,其ensembl ID如下:
"ENSG00000242268" "ENSG00000270112" "ENSG00000167578"
"ENSG00000273842" "ENSG00000078237" "ENSG00000146083"
想要将他们转换成对应的基因名字,即hgnc name,代码如下:
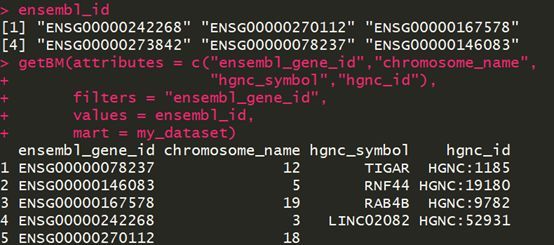
大家首先看代码,再来看结果。代码中,attributes参数定义了四个输出项,分别是ensembl_gene_id,chromosome_name, hgnc_suymbol以及hgnc_id,所以我们的输出有四列。而filters参数定义的是我们的输入项,由于此处我们的原始信息是ensembl_gene_id,因此此处很显然就定义成ensembl_gene_id;values参数接受我们的原始信息;mart参数定义数据集。
大家再来看结果,我们明明输入了6个ensembl_gene_id,但是却偏偏只出来五行结果,ENSG00000273842被漏掉了?还有一个问题,ENSG00000270112怎么没有对应的hgnc信息?其实大家不必惊慌,首先明白一点,并非所有的ensembl_gene_id都有对应的hgnc信息,这是很正常的;其次,由于数据库的更新,某些老旧的ensembl_gene_id会被遗弃,比如这里的ENSG00000273842!
其余基因ID的转换如出一辙,在此不做过多介绍。
四、查看富集到某个GO ID上的基因
我们也可以根据某个GO ID,查看富集到该通路上的基因,比如:
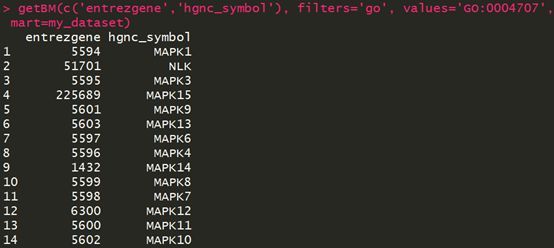
此处返回的是基因的entrez ID和hgnc_symbol,输入是GO:0004707,最后得到共有14个富集在此通路上的基因。当然我们也可以对输出的基因进行限定,比如:
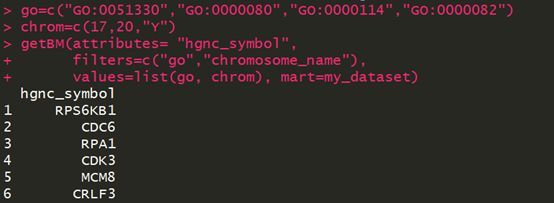
此处我们设定了4个GO ID,然后增加了一个限定条件,就是这些基因必须出现在第17或者第20条或者Y染色体上,最终仅有6个基因返回。
五、对基因进行GO注释
与第四步相反,我们可以根据基因的某个ID,查看它在GO富集中的ID号,比如:
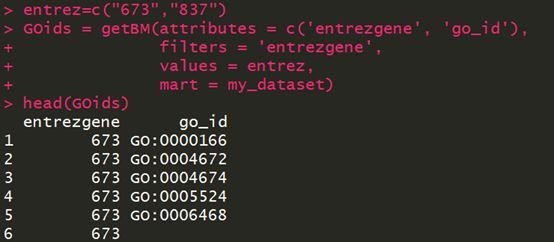

好了,以上就是关于biomaRt包中关于基因ID转换的内容。当然此处只是给大家抛砖引玉,基因ID很多,这个包的功能也很多,但是基本的思想和套路是相似的,大家可以从自己最熟悉的ID入手,尝试几次之后,就可以彻底掌握biomaRt的用法了。不过在这之前,Leopard还是建议大家先搞清楚不同的基因ID到底是怎么来的,至少得知道是谁命名的。
- 本文固定链接: https://maimengkong.com/kyjc/930.html
- 转载请注明: : 萌小白 2022年5月21日 于 卖萌控的博客 发表
- 百度已收录
