为什么要做调研图
基因组测序现在已经成为生物学研究的一个重要手段,基因组的杂合度和重复序列对后续基因组组装有很大的影响。高杂合的基因组往往无法合并姊妹染色体,导致组装的结果偏大,而重复序列在组装中会被折叠,使组装中出现缺口、错误,导致组装的结果偏小。不同的生物体的基因组之间杂合率和重复序列含量差异巨大,因此在进行基因组测序前往往需要对基因组的特征进行调研,以确定测序方案,周期等。目前常用的调研手段有三种:
- 用流式细胞仪测定细胞核内的 DNA 总量
-
- 用调研图,通过二代测序,估算基因组大小、杂合度、重复序列比例、 GC 含量等。
不同的技术手段有不同的侧重,其中调研图以低成本,低难度和更多的评估内容成为使用最多的技术手段,同时调研图所测的二代数据还可以用于回比基因组,以评估组装质量。
调研图是基于数学统计学手段获取物种信息的方式,因此对于已经研究的较为清晰的物种—— 主要是普通二倍体和简单多倍体,其染色体条数、倍性、大概基因组大小是已知的,此时仅选择调研图就足以满足了解基因组特征的需要,但对于多倍体复杂基因组更推荐补充核型分析和流式的结果,以和调研图相互印证补充。
调研图原理
(以二倍体调研图为例)
评估基因组大小
调研图最重要的用处是对基因组的大小进行估计。,对于没有测序错误的理想情况下,用测序数据估算基因组大小可以按照这个公式:基因组大小= 测序量/reads平均覆盖深度。但由于测序错误的存在,较长的reads 非常容易受到错误的影响,而将reads切碎为“ 长度为k 的片段” ,即k-mer ,能大大减轻这个问题,因此调研图绘制的是k-mer 的深度-频率分布图。此时估算基因组大小的公式就变成了:基因组大小= 正常kmer数量/k-mer平均覆盖深度。因为错误总是随机出现的,所以这里正常的k-mer 数是过滤掉过低频率的k-mer(即错误)后得到的数量。
在理想状态下,K-mer 曲线服从泊松分布,即只会出现一个明显的主峰。但对于一个杂合二倍体,主峰前1/2 出会出现一个杂合峰,在杂合度较高的时候可能出现高过主峰的情况。下图即为一个高杂合二倍体kmer 的频率-深度分布图。主峰后二倍位置内的峰为重复峰。但如果杂合度很低,可能分布图中只有一个峰存在。主峰所代表的就是k-mer 平均覆盖深度,用主峰深度代替公式中的k-mer 平均覆盖深度即可算得该基因组的大小。
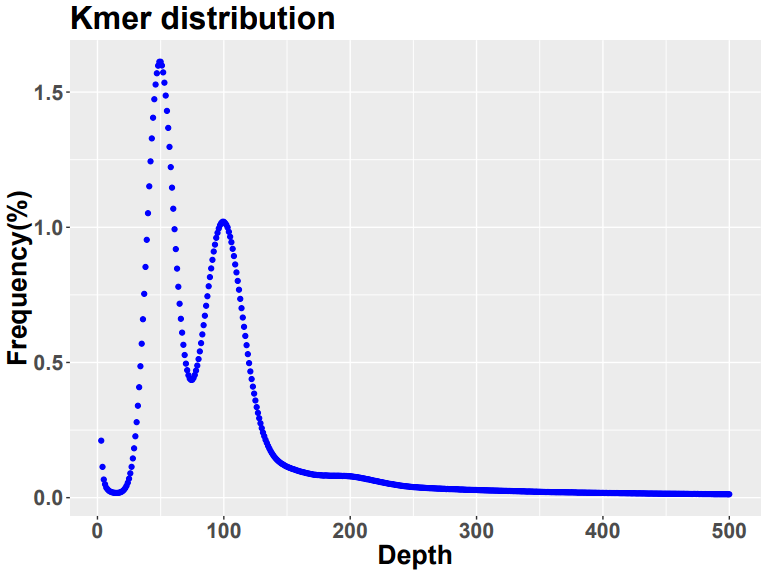
Figure 1,一个二倍体的kmer 频率- 深度分布图,横轴为深度,纵轴为kmer 出现的频率,主峰位于深度100 左右,杂合峰位于深度50 左右
k-mer 也并不是切的越小越好,过短的k-mer将无法保证多数k-mer在基因组中只出现一次,导致主峰深度估计偏大,而较长的k-mer 具有跨越更长重复片段的能力,因此k-mer 的选择其实是一个平衡错误和重复的过程。通常k 的选择为15 到21的奇数,既能够保证k -mer的种类能覆盖基因组,又足够小以避免错误的影响。基因组中往往还存在一些重复序列,这些重复序列也会引起kmer 的重复,但这些重复的存在虽然会削低主峰的高度,却不会改变主峰的位置,上图主峰后的小峰即为重复峰。
评估杂合率和重复序列
前面已经提到了杂合率的高低对基因组的组装有非常大的影响,那么如何计算基因组的杂合率和重复率呢?通常有两种不同的方法,一种是直接计算峰的面积,即上图中杂合峰和重复峰的面积占总面积的比例,从而估算出杂合率和重复率。另一种是通过模型拟合各个峰。
genomescope21就是一个通过负二项分布拟合基因组k-mer 分布来评估基因组特征的工具,其结果被普遍认可。以下图为例,一个二倍体的genomeScope 分析结果。
- 蓝色 柱子是 kmer 的观测值;
- 橙红色 拟合线部分对应着深度过低的 kmer ,这些 kmer 被认为是测序错误引入的;
- 黑色 拟合线是除去被认为是错误的部分(橙红色拟合线部分)之后剩下的所有 k-mer ,这些被认为是可靠的 kmer 数据;
- 黄色 拟合线被认为来自基因组非重复区域的 K-mer 分布;
- 垂直的黑色虚线 为预测最低深度峰的整数倍覆盖度;
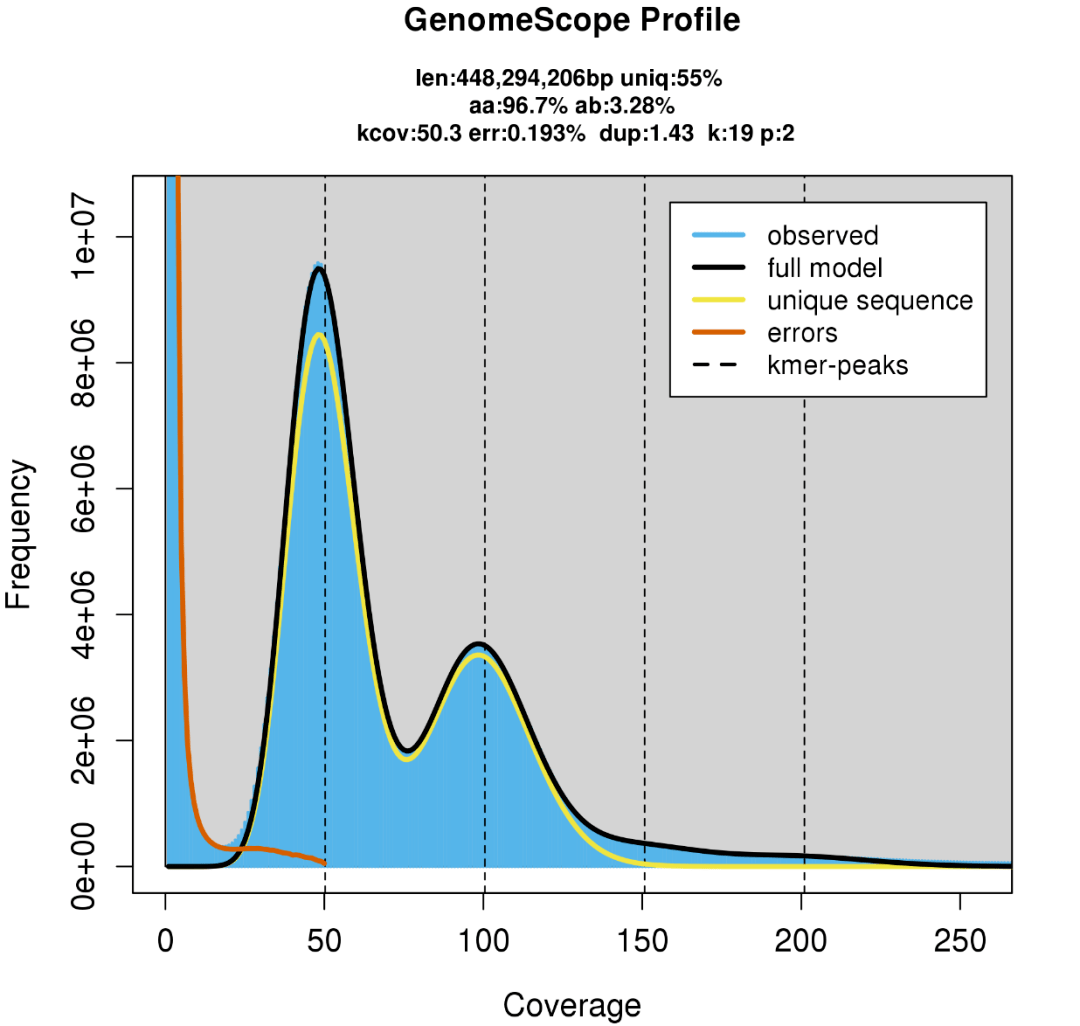
Figure 2一个二倍体的调研图,横轴是测序深度( 覆盖度),纵轴是k-mer 出现的频率,主峰位于100 左右。
多倍体调研图
多倍体又分为异源多倍体和同源多倍体,其调研图的情况更为复杂。以四倍体为例,异源四倍体又被称为双二倍体,从k-mer 分析的角度来说,其调研图和二倍体并无太大差异。虽然如此,他们的染色体之间仍然存在一定的相似性,所以在主峰二倍的位置上往往存在一个小的凸起,这样的凸起和高重复率的二倍体调研图非常接近。如下图是一个异源四倍体的调研图:
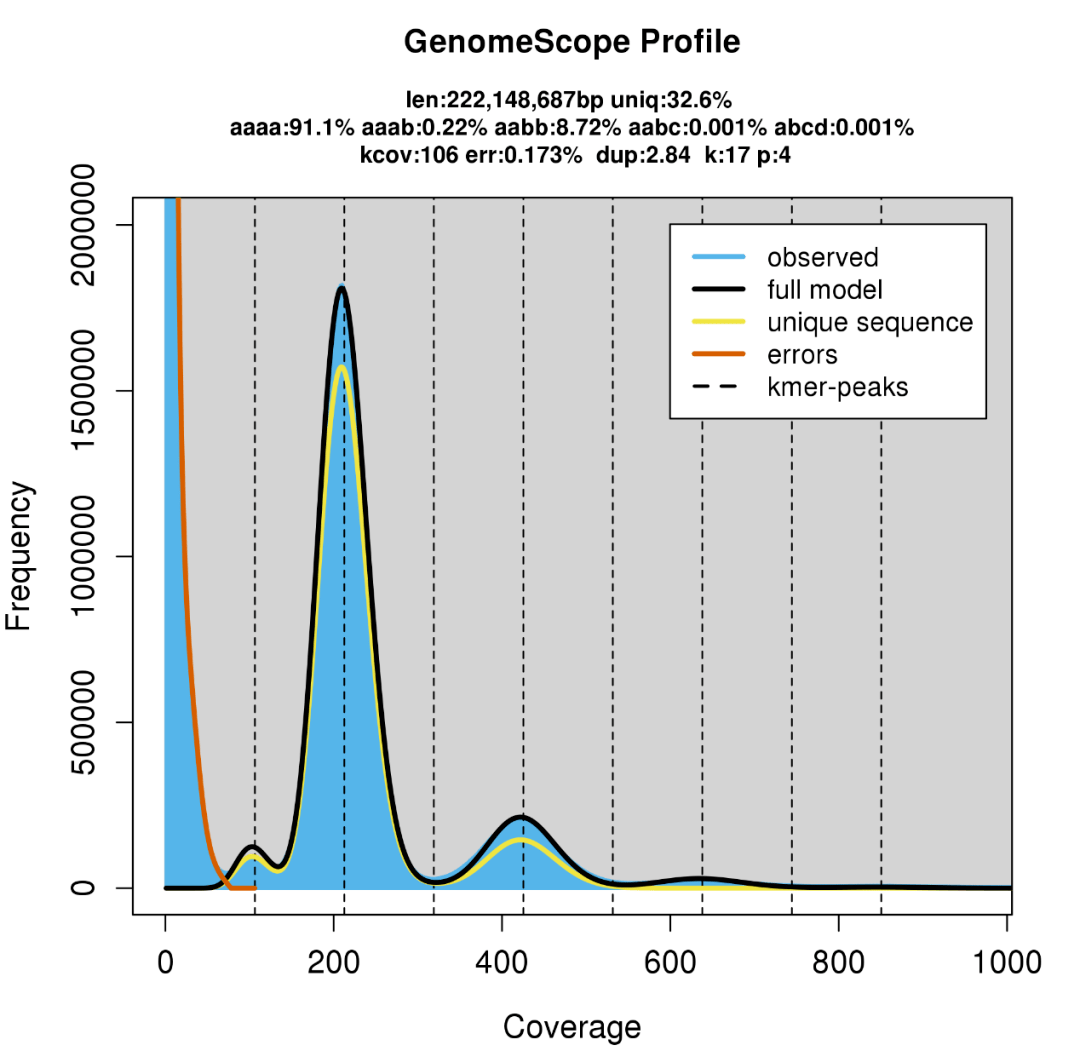
Figure 3一个异源四倍体的调研图,主峰位于212 深度,三个峰的比例为1:2:4
同源四倍体的两套亚基因组之间的区别比异源四倍体更为接近,体现在调研图上就是在主峰的二倍位置处有一个明显隆起的峰。如果存在一定的杂合率,调研图上就会存在三个比例为1:2:4 的峰,但如果基因组的杂合率很低,杂合峰不明显,此时调研图看起来和二倍体仍然非常接近。
不同的倍性对整套基因组的大小影响不大,所以面对多倍体时,调研图的结果最好同时结合流式细胞仪或者核型来判断。对于杂合率约在0.5%~20% 左右,重复序列不超过约40% 的物种,也可以利用smudgeplot软件1对杂合k-mer 进行分析,得到可能的物种倍性,从而辅助基因组杂合率和重复序列的估计。
Smudgeplot 通过寻找杂合k-mer 来研究基因组的倍性,其定义的杂合k-mer 对指的是一对k-mer 之间只相差一个碱基,且没有第三个k-mer与他们再相差一个碱基(如ATGATCA, ATGCTCA, ATGGTCA)。对于一个AB 形式的杂合,smudgeplot 试图从所有k-mer 中寻找一对杂合k-mer,而对于一个AAB 形式的杂合,smudgeplot 试图寻找两条相同的k-mer 和一条它们的杂合k-mer,在图中表示为更高的亮度。如下图对一个四倍体的smudgeplot 分析,可以明显的看出AABB 杂合模式附近的k-mer 数量明显高于其它k-mer,左上角也描述了这个结果。
Figure 4一个四倍体的smudgeplot 分析结果,横轴是杂合k-mer对的深度占总深度的比例,纵轴是所有k-mer对的总深度,二者的交点即代表了不同的杂合结构。交点的亮度代表了落入其中的k-mer数。
需要注意的是,调研图一般需要50x 以上的数据对基因组进行评估,在测序深度不足的情况下,调研图可能无法很好的形成真正意义上的峰,此时最好的选择是加测数据,重新绘制调研图进行评估。
[1] Ranallo-Benavidez, T.R., Jaron, K.S. & Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun11,1432 (2020).
转自:百迈克基因
- 本文固定链接: https://maimengkong.com/kyjc/744.html
- 转载请注明: : 萌小白 2021年8月13日 于 卖萌控的博客 发表
- 百度已收录
