大家好,这是专注表观组学十余年,领跑多组学科研服务的易基因。
本期,我们讲讲cfDNA重亚硫酸盐测序(cfDNA-RBS)实验怎么做,从技术原理、建库测序流程、信息分析流程等方面详细介绍。
一、cfDNA重亚硫酸盐测序(cfDNA-RBS)技术概述
在⼈类基因组中,90%以上的CpG位点是被甲基化的,CpG位点的分布有两种形式,⼀种是少量分散在基因组中,另⼀种就是集中在启动⼦周围⾼密度的CpG岛。DNA甲基化参与各种不同的基因调控过程,从⽽调控多种复杂的⽣物过程,如胚胎发育,衰⽼和肿瘤发⽣等,哺乳动物CpG甲基化有助于基因组的稳定性和转录调控。除启动⼦外,远端调控元件的DNA甲基化模式与细胞特性和染⾊质结构相关,特别是在增强⼦和CTCF结合位点,因此全⾯分析DNA甲基化对了解细胞状态和动态⾄关重要。DNA甲基化的⽣物学作⽤已通过全基因组或简化基因组甲基化测序分析⽅法阐明,但这些⽅法不能有效地研究哺乳动物基因组中的⼤量⾮编码调控元件。全基因组甲基化测序(WGBS)是覆盖整个基因组范围,费⽤较⾼,效率相对较低。⽽简化甲基化测(RRBS)主要针对CpG富集区域进⾏甲基化测序,缺乏CpG岛以外的增强⼦区域和CTCF结合位点的覆盖。因此研究⼈员提出了⼀种可以富集覆盖启动⼦、增强⼦、CTCF结合位点的甲基化测序⽅法——cfDNA-reduced representation bisulfite sequencing, cfDNA-RBS,该⽅法可以在低样本量和单细胞中⾼效兼容。
cfDNA-RBS的优势:
(1)cfDNA-RBS覆盖更⼴泛的基因调控元件,且具有更⾼的效率和更低的测序深度求。
(2)cfDNA-RBS可检测不同细胞类型的DNA甲基化差异。
(3)cfDNA-RBS可预测功能元件的状态。
(4)cfDNA-RBS 可应⽤于单细胞DNA甲基化研究。
cfDNA-RBS测序原理示意图如下:
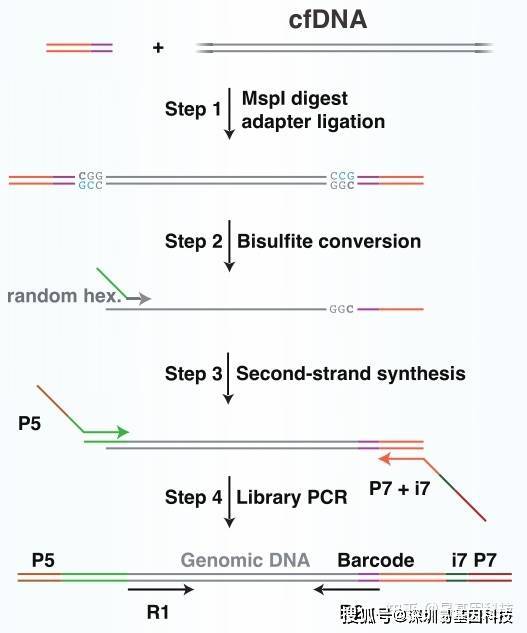
二、cfDNA重亚硫酸盐测序(cfDNA-RBS)建库测序
(一)DNA提取与检测
从组织或细胞中提取DNA,随后对DNA样品进⾏严格质控,质控标准主要包括以下⼏个⽅⾯:琼脂糖凝胶电泳:分析样品DNA完整性及是否存在降解;Qubit fluorometer( Quant-iT dsDNA HS Assay Kit):检测DNA总量。
(二)⽂库构建与质检
(1)文库构建:
检测合格后,取 5-10ng 基因组 DNA 加⼊ Msp I 酶在 37℃条件下消化 3h。纯化后的⽚段 DNA ⽤ T4 DNA 连接酶将 5 ' -甲基胞嘧啶合成的接头连接起来。纯化后,进⾏Bisulfite 处理(采⽤EZ DNA Methylation Gold Kit, Zymo Research),经过处理,未发⽣甲基化的C变成U,再通过Random hexame primer进⾏扩增和PCR扩增后,得到最终的⽂库。

(2)文库质检:
⽂库构建完成后,先使⽤ Qubit2.0 进⾏初步定量,稀释⽂库⾄ 1ng/μl,随后使⽤ Agilent 2100 对⽂库的 insert size 进⾏检测,insert size 符合预期后,使⽤qPCR ⽅法对⽂库的有效浓度进⾏准确定量(⽂库有效浓度> 2nM),以保证⽂库质量。
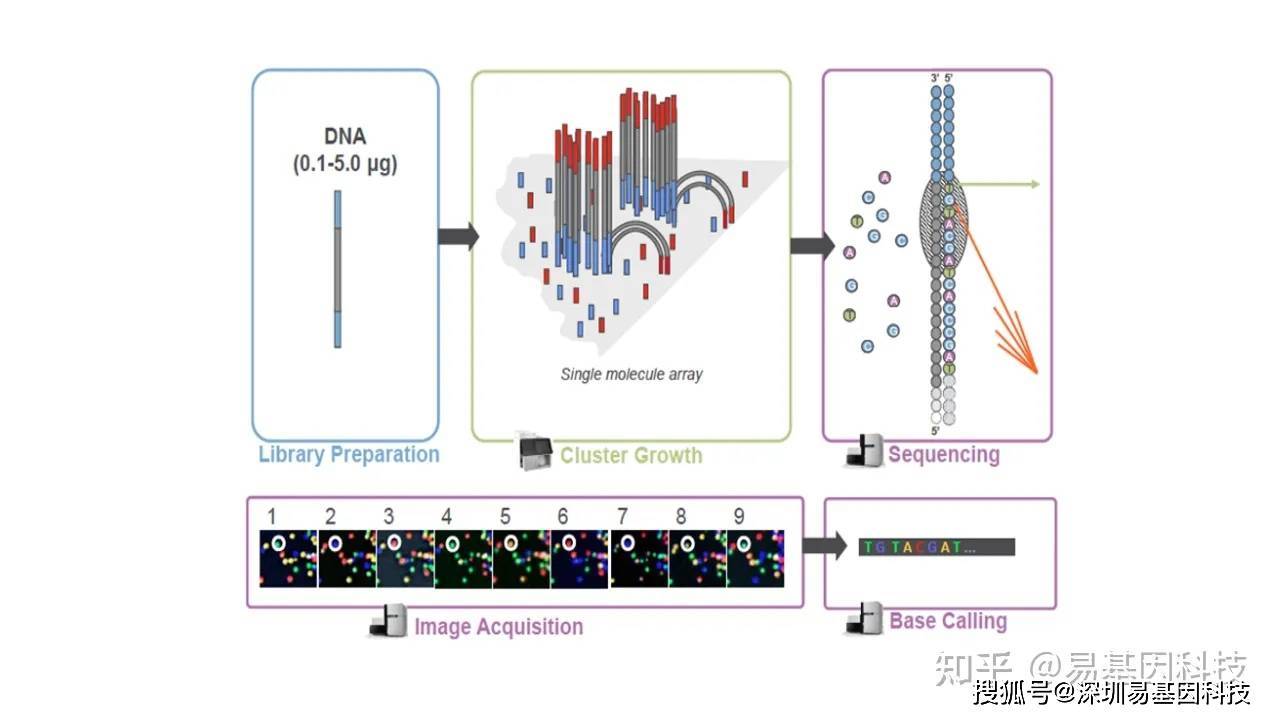
(三)上机测序
库检合格后,把不同⽂库按照有效浓度及⽬标下机数据量的需求pooling后进⾏Illumina测序。测序过程如下图所示。
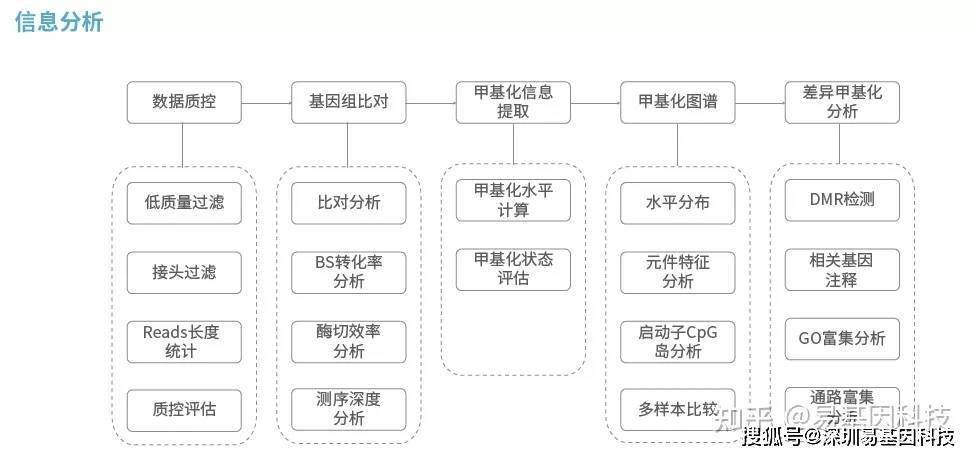
三、cfDNA重亚硫酸盐测序(cfDNA-RBS)信息分析流程
将测序结果与参考基因组⽐对,⽐对上唯⼀位置的序列⽤于后续标准信息分析及个性化分析。信息分析流程如下:
四、cfDNA重亚硫酸盐测序(cfDNA-RBS)质控分析
(一)测序数据说明
测序⽚段被⾼通量测序仪测得的图像数据经CASAVA碱基识别转化为序列数据(reads),⽂件为fastq 格式,其中主要包含测序⽚段的序列信息以及其对应的测序质量信息。fastq格式⽂件中每个read由四⾏描述信息组成,如下所示:
 图:FASTQ格式示例
图:FASTQ格式示例
上述⽂件中第⼀⾏以“@”开头,随后为Illumina测序标识符(Squence Identifiers)和描述⽂字;第⼆⾏是测序⽚段的碱基序列;第三⾏以“+”开头,随后为Illumina测序标识符(也可为空);第四⾏是测序⽚段每个碱基相对应的测序质量值,该⾏中每个字符对应的ASCII值减去33,即为该碱基的测序质量值。

测序过程本身存在发⽣机器错误的可能性,测序错误率分布检查可以反映测序数据的质量,序列信息中每个碱基的测序质量值保存在FASTQ⽂件中。如果测序错误率⽤e表示,Illumina的碱基质量值⽤Q 表示,则有:Q =-10log10(e)。Illumina Casava 1.8版本碱基识别与Phred分值之间的简明对应关系⻅下表:
(二)测序数据质控
原始下机数据包含建库时引进的接头序列以及质量过低的碱基,这些因素会导致后续⽐对到基因组的reads较少,从⽽导致得到的信息较少,因此需要进⾏过滤。针对cfDNA-RBS⽂库结构的特殊性,对引⼊的随机引物进⾏去除。
(三)数据质量评估
经过原始数据过滤、测序错误率检查、GC含量分布检查。
(四) ⽐对质量评估
经过质控的reads需要根据与参考基因组的序列相似度⽐对到参考基因组上。
相⽐于常规基因组及转录组测序,cfDNA-RBS测序⽅法产⽣的数据的特点决定其在⽐对时存在三⼤困难:
(1)DNA⽚段正链和负链经过重亚硫酸盐转化后将不再反向互补,再经过PCR,便会产⽣四条不同的序列,这将⼤⼤增加⽐对时的计算量。
(2)经过重亚硫酸盐转化后,DNA序列⼤部分C碱基被转化成T碱基,因此序列含⼤量T⽽缺乏C;经过PCR后,产⽣的互补链则含有⼤量A⽽缺乏G。这样便导致序列的复杂度降低(即序列的组成特征更单⼀),从⽽增加⽐对的难度。
(3)C和T的⽐对是不对称的。经过重亚硫酸盐转化后,序列中⾮甲基化的C碱基(占⼤部分)被转化为T,这将导致测序序列与参考基因组不匹配,T既可能应该⽐对到T上,有可能应该⽐对到C上;⽽C则只能⽐对到C上。这也增加了⽐对的难度。
经过质控的reads根据与参考基因组的序列相似度⽐对到参考基因组上。⽐对到参考基因组上的reads数量越多,后续分析过程中可利⽤的信息就越多。
(五)转化效率及覆盖率评估
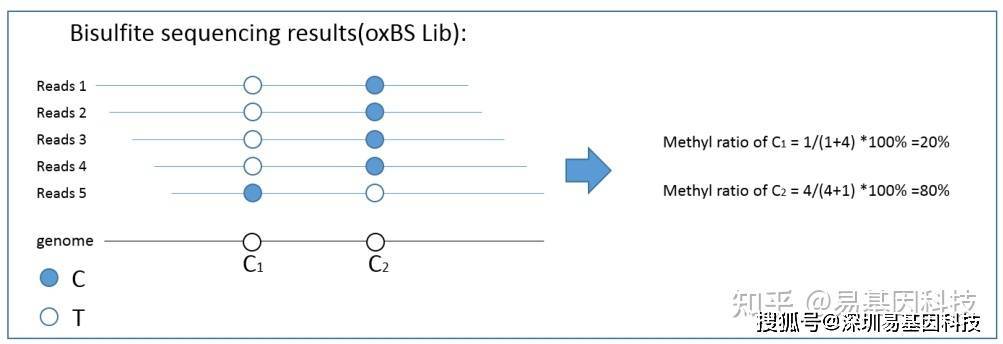
(1)甲基化⽔平计算
甲基化⽔平可根据未转化为 T 的 C 与转化为 T 的 C 的 reads 的⽐例计算得到,即:
Beta-value = C-reads / (C-reads + T-reads) * 100%
其中,Beta-value 即为该胞嘧啶的甲基化⽔平,C-reads 为覆盖该位点的⽀持甲基化的reads 数⽬(测得该位点为 C 的reads),T-reads 为覆盖该位点的不⽀持甲基化的 reads 数⽬(测得该位点为 T 的 reads)。 计算原理示意图如下:
(2)转化效率评估
对于cfDNA-RBS⽂库,经过重亚硫酸盐的转化,⽂库中⾮甲基化的C可转化为T。在建库时加⼊lambda序列可以对⽂库的转化效率进⾏评估。
(3)覆盖率评估
将reads⽐对到基因组后,⽐对到不同位点的reads数(测序深度)不同,测序深度过低会导致计算的甲基化率不可信。因此需要统计所有C位点的测序深度。
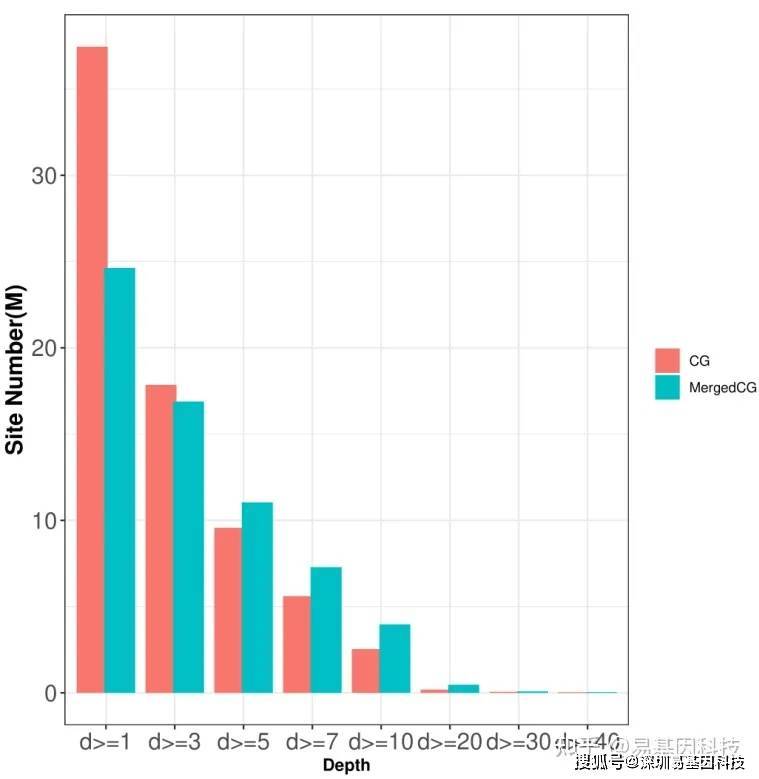
五、cfDNA-RBS基因组酶切富集⽚段区域甲基化⽔平概览
鉴定每个样本基因组酶切富集⽚段区域甲基化⽔平,并从不同⽅⾯进⾏统计、展示,以从宏观⽔平了解每个样本的甲基化⽔平。
(1)样本基因组酶切富集⽚段区域甲基化图谱
(2)甲基化平均⽔平统计
分别统计有效测序深度下,三种类型胞嘧啶基因组酶切富集⽚段区域范围内甲基化的平均⽔平。该统计可以从宏观⽔平反应样本基因组酶切富集⽚段区域甲基化⽔平的⾼低。
(3)基因组酶切富集⽚段区域甲基化的分布
不同类型的胞嘧啶的甲基化⽔平在不同物种间,甚⾄同⼀物种不同细胞类型间都存在差异。因此,统计了每种类型C碱基甲基化⽔平的分布。
将CG、CHG、CHH类型的胞嘧啶,按照甲基化⽔平划分为10个档次(0-0.1, 0.1-0.2, 0.2-0.3等),分别统计每个档次包含的胞嘧啶的⽐例。
(4)各样本甲基化⽔平分布⽐较
展示各样本基因组酶切富集⽚段区域甲基化⽔平在不同区间的的分布密度,直观地⽐较样本间甲基化⽔平分布的特征。
(5)各基因元件的甲基化⽔平特征
不同基因元件的甲基化⽔平不同。将基因组序列按照不同的基因结构元件划分为启动⼦(promoter),基因本体(genebody),转录终⽌位点下游2000bp(down2k)并统计这些基因结构元件中,MergedCG位点的甲基化⽔平的分布。
(6)基于基因组酶切富集⽚段区域甲基化图谱的分层聚类
根据基因组酶切富集⽚段区域甲基化图谱图谱,对样本进⾏聚类,可从总体⽔平反应样本组内和组间差异。
(7)基于基因组酶切富集⽚段区域甲基化图谱的PCA分析
主成分分析(PCA)是⼀种常⽤的数据降维⽅法,其⽬标是寻找⼀组新的变量,使它们反映原始数据的主要特征。每个新变量是原有变量的线性组合,称为“主成分”。这组新变量中,只有少数⼏组变量能够反映原有变量的主要特征,通过这种⽅式,可以使原有数据的维度降低,更易于体现各样本的特征。
六、cfDNA-RBS差异甲基化位点(DMC)的鉴定及统计
(1)差异甲基化位点(DMC)的鉴定
(2)DMC的注释
鉴定出的DMC包含染⾊体、起始位置、终⽌位置等信息。根据DMC的位置信息,结合基因组注释信息中所有基因的位置信息及各个基因元件(up2k,promoter, 5utr, cds, exon, intron, 3utr, genebody, down2k, intergenic, cgi, cgi_up4k, cgi_down4k)等位置信息,鉴定DMC与哪些基因的哪些基因元件有重叠,以此来判断DMC修饰哪些基因的哪些基因元件。
(3)DMC修饰基因的统计
根据DMC的注释⽂件,提取出DMC修饰的基因及其的信息(DMC位于基因的promoter或genebody区时,认为该DMC修饰该基因),以更加⽅便地查看DMC修饰的基因。
(4)DMC在染⾊体上的分布
根据DMC的位置信息,统计DMC落在哪些染⾊体上,并⽤图形展示,以了解DMC在染⾊体上的分布有偏好性。
(5)DMC在基因元件上的分布
根据DMC的位置信息,分别统计Hyper DMC及Hypo DMC 落在哪些基因元件上
(6)DMC修饰基因的功能富集分析
基因本体( Gene Ontology, GO)是基因功能国际标准分类体系,提供了⼀套动态更新的标准词汇表来描述⽣物体中基因和基因产物的属性,可以挖掘出⼀些⽣物学相关的途径。 GO分为三个Ontology,分别是:分⼦功能(Molecular Function, MF)、细胞组分( Cellular Component, CC)和⽣物过程(Biological Process, BP)。
KEGG( Kyoto Encyclopedia of Genes and Genomes,京都基因与基因组百科全书)是基因组破译⽅⾯的数据库。在给出染⾊体中⼀套完整基因的情况下,它可以对蛋⽩质交互⽹络在各种细胞活动起的作⽤做出预测。KEGG Pathway显著性富集分析应⽤超⼏何检验,找出与整个基因组背景相⽐,在差异甲基化修饰的基因中显著富集的Pathway。
将鉴定出的DMC所修饰的基因,利⽤GO和KEGG数据库进⾏功能富集分析。将DMC修饰基因进⾏三种分类,并对每个分类做富集分析:
⾸先,对Hyper-DMC、Hypo-DMC修饰的基因(⽆论修饰位于genebody还是promoter)分别做功能富集分析。
⽬前⼤部分报道公认若甲基化修饰位于基因的promoter区,会抑制基因的表达;⽽甲基化修饰位于genebody区时,其对基因表达的影响则更加复杂,有报道称genebody区甲基化⽔平与表达呈正相关,但两者之间的关系实际上根据细胞的不同⽽不同;羟甲基化与基因表达之间的关系研究的更少。有报道认为,TSS附近的区域(TSS上游临近区域及第⼀个启动⼦)羟甲基化⽔平与表达呈负相关,genebody区羟甲基化⽔平则与表达呈正相关。因此,对位于promoter区及genebody区的Hyper-DMC和Hypo-DMC修饰的基因也分别进⾏富集分析。
综上所述,对DMC来说,都对以下四类基因进⾏功能富集分析:
- 对实验组相对于对照组甲基化⽔平升⾼的DMC(All-Hyper DMC)修饰的基因做功能富集分析;对实验组相对于对照组甲基化⽔平降低的DMC(All-Hypo DMC)修饰的基因做功能富集分析;对实验组相对于对照组甲基化⽔平升⾼的启动⼦区的DMC(Promoter-Hyper DMC)修饰的基因做功能富集分析;对实验组相对于对照组甲基化⽔平降低的启动⼦区的DMC(Promoter-Hypo DMC)修饰的基因做功能富集分析。
富集分析采⽤Fisher检验,结合BH矫正。富集分析结果包括表格和图⽚两部分,其中,表格为所有富集到的GO/KEGG条⽬,包括显著和不显著的。
七、cfDNA-RBS差异甲基化区域(DMR)的鉴定及统计
(1)差异甲基化区域(DMR)的鉴定
(2)DMR的注释
鉴定出的DMR包含染⾊体、起始位置、终⽌位置等信息。根据DMR的位置信息,结合基因组注释信息中所有基因的位置信息及各个基因元件(up2k,promoter, 5utr, cds, exon, intron, 3utr, genebody, down2k, intergenic, cgi, cgi_up4k, cgi_down4k)等位置信息,鉴定DMR与哪些基因的哪些基因元件有重叠,以此来判断DMC修饰哪些基因的哪些基因元件。
(3)DMR修饰基因的统计
根据DMR的注释⽂件,提取出DMR修饰的基因及其的信息(DMR位于基因的promoter或genebody区时,认为该DMC修饰该基因),以更加⽅便地查看DMR修饰的基因。
(4)DMR在染⾊体上的分布
根据DMR的位置信息,统计DMR落在哪些染⾊体上,并⽤图形展示,以了解DMR在染⾊体上的分布有偏好性。
(5)DMR在基因元件上的分布
根据DMR的位置信息,分别统计Hyper DMR及Hypo DMR 落在哪些基因元件上。
(6)DMR修饰基因的功能富集分析
同样地,对DMR修饰的基因也分成四类做功能富集分析。
- 对实验组相对于对照组甲基化⽔平升⾼的DMR(All-Hyper DMR)修饰的基因做功能富集分析;对实验组相对于对照组甲基化⽔平降低的DMR(All-Hypo DMR)修饰的基因做功能富集分析;对实验组相对于对照组甲基化⽔平升⾼的启动⼦区的DMR(Promoter-Hyper DMR)修饰的基因做功能富集分析;对实验组相对于对照组甲基化⽔平降低的启动⼦区的DMR(Promoter-Hypo DMR)修饰的基因做功能富集分析;
富集分析采⽤Fisher检验,结合BH矫正。富集分析结果包括表格和图⽚两部分,其中,表格为所有富集到的GO/KEGG条⽬,包括显著和不显著的。
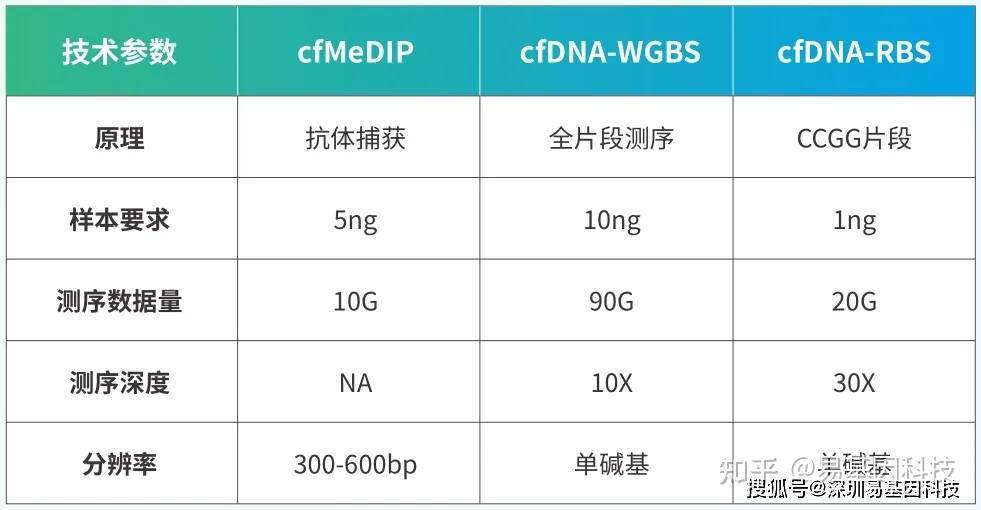
易基因微量cfDNA甲基化测序(cfDNA-BS)技术介绍
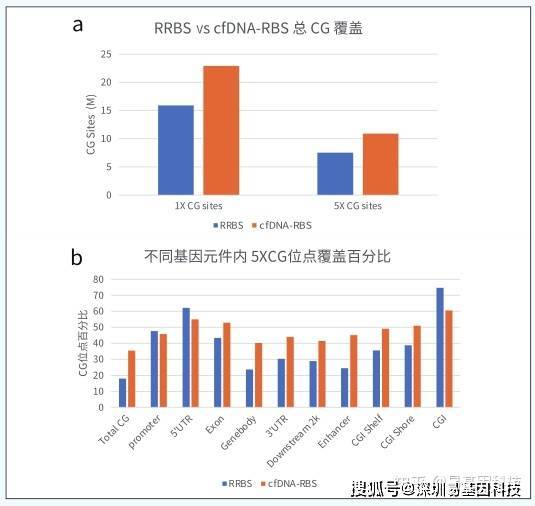
cfDNA片段化严重,片段大小常在150bp左右,现有甲基化检测技术包括cfMeDIP和微量WGBS等。无法做到碱基分辨、具有抗体特异性和非特异性捕获、覆盖深度低、检测成本高等特点。常规RRBS富集约70-350bp范围酶切片段,如对于CG含量高的片段将被切割的更碎而无法检测,保留下来的片段反而是CG含量低,无甲基化信息的基因片段。
易基因研发cfDNA-RBS技术,特异性捕获CCGG位点两端的DNA,通过亚硫酸盐测序,实现高深度,单碱基分辨检测CG位点甲基化信息。DNA起始量仅需1ng,是目前肿瘤甲基化标志物检测研究的优选技术。
技术优势:
- 超低起始量:100-500ul血浆或1ng cfDNA;测序覆盖度高:20G测序数据,可达10M的CG位点覆盖,涵盖CpG岛、启动子、增强子、CTCF结合位点等多种核心调控区域单碱基分辨率:在其覆盖范围内可精确分析每一个C碱基的甲基化状态;性价比高:成本相对于现有技术大幅降低。
技术指标:
应用场景:
- 癌前病变的癌变预警标志物检测肿瘤早期筛查标志物检测肿瘤预后标志物检测药物疗效预测标志物检测
以上就是关于cfDNA甲基化测序实验流程和分析思路的介绍,易基因科技提供全面的DNA甲基化研究整体解决方案,技术详情了解请致电易基因0755-28317900。
参考文献:
[1] Ashburner, M. and C. A. Ball, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet, 2000, 25 (1): 25-9.
[2] Dirk Schübeler. Function and information content of DNA methylation. Nature, 2015, 517: 321–326.
[3] Frank Jühling et al. metilene: Fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Research, 2016, 26: 256-262.
[4] Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research, 2000,28(1): 27-30.
[5] Tadafumi Kato Kazuya Iwamoto. Comprehensive DNA methylation and hydroxymethylation analysis in the human brain and its implication in mental disorders.
Neuropharmacology, 2014, 80: 133-139.
[6] Xiaojing Yang et al. Gene Body Methylation Can Alter Gene Expression and Is a Therapeutic Target in Cancer. Cancer Cell 26, 577–590.
[7] Yuanxin Xi et al. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics, 2009, 10:232.
[8] Shareef, S.J., Bevill, S.M., Raman, A.T. et al. Extended-representation bisulfite sequencing of gene regulatory elements in multiplexed samples and single cells.
Nat Biotechnol 39, 1086–1094 (2021).

转自:易基因
- 本文固定链接: https://maimengkong.com/kyjc/1233.html
- 转载请注明: : 萌小白 2022年10月4日 于 卖萌控的博客 发表
- 百度已收录
