一、介绍
宏基因组 ( Metagenome) 指特定环境下所有生物遗传物质的总和。它包含了可培养的和未可培养的微生物的基因。一般从环境样品中提取基因组DNA, 进行高通量测序,从而分析微生物多样性、种群结构、功能信息、与环境之间的关系等。
宏基因组的分析目前主要包括三种方法:基于组装分析、基于reads分析、基于bin分析。
下面我们介绍基于bin的分析方法。
二、分析流程介绍
宏基因组分箱(Binning)是将序列组装得到的Contigs按物种分开归类的过程。宏基因组分箱技术有助于获得不可培养微生物的全基因组序列,获得新物种的基因组序列和功能,预测未知物种的培养方法等等。
分箱软件通常基于GC含量、核苷酸频率、关键的单拷贝基因序列等组成性特征;丰度分布差异,认为源自同一基因组的序列 (contigs),在同一样本中应具有一致的丰度,而在不同的样本之间,它们又应该具有相似的丰度分布;两种策略进行分箱。
基于Binning的宏基因组分析流程,数据分析从下机原始序列开始,首先对原始序列进行去接头、 质量剪切以及去除污染等优化处理。然后使用优质序列进行拼接组装得到Contigs;使用metabat2和maxbin2软件分别对每个样本的Contigs进行分箱;不同软件分箱得到的bin进行合并(binning_refiner)、提纯(MAGpurify);然后将所有样优化后的bin进行去冗余(dRep);而后分别从物种和功能方面进行信息统计。
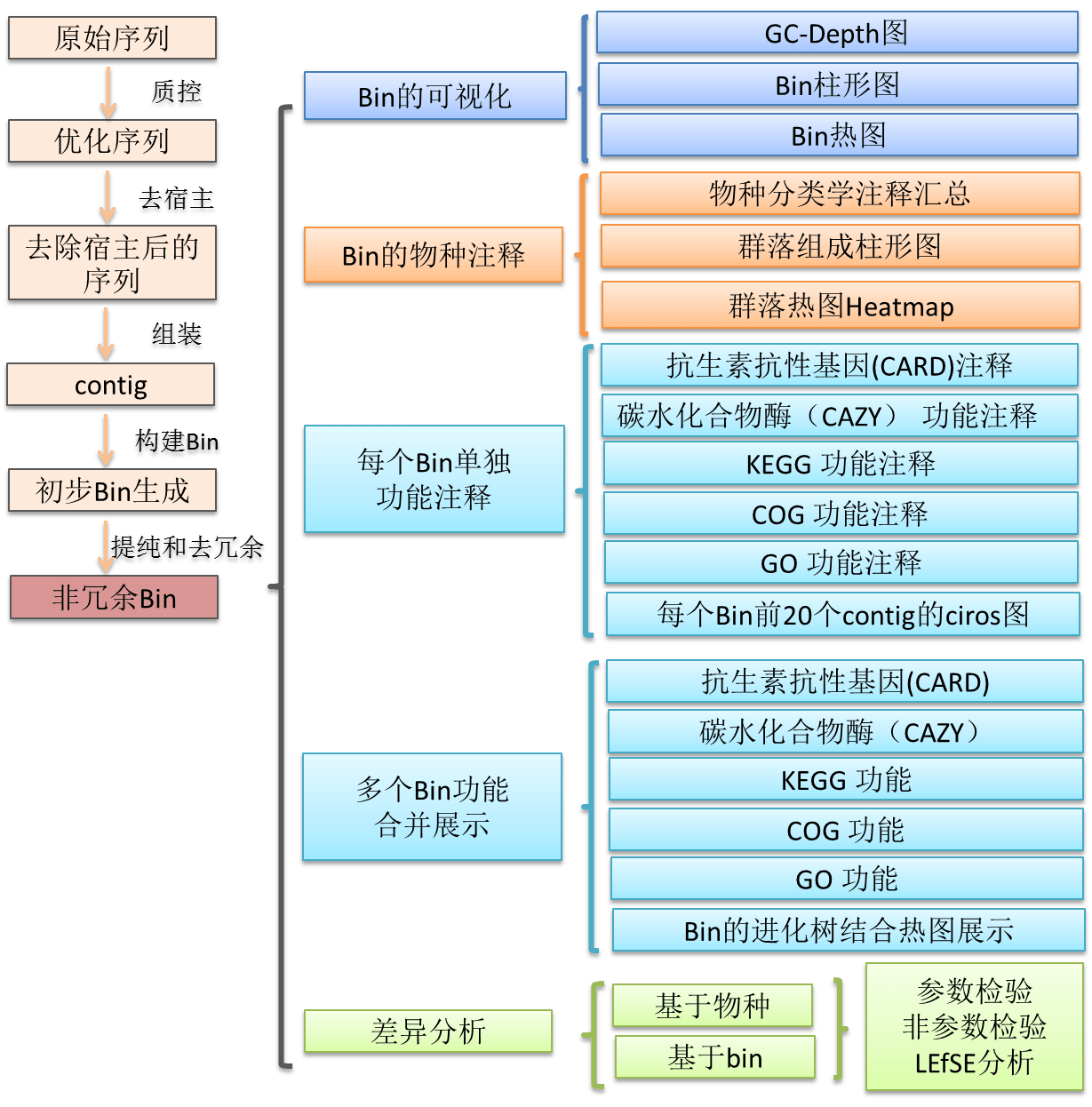
三、详细流程
1. 数据质量控制,为了提高后续分析质量和可靠性,对原始测序数据进行以下清洗处理, 获取用于后续分析的有效数据(Clean Data)。具体处理步骤如下:使用fastp软件去除质量低的reads,过滤掉质控后长度较低的reads,同时去掉接头序列;如果样品来源于宿主(比如人或动物的粪便),而且该宿主本身的基因组已被发表, 则通过软件Bowtie2软件将reads比对宿主DNA序列,并去除比对相似性高的污染reads;
2. 序列组装: 使用Megahit软件使用不同kmer对优化序列进行组装得到Contigs;
3. Bin生成:采用metaWRAP环境中binning模块的metabat2和maxbin2方法对contig进行分箱。
4. 合并不同软件得到的bin:采用binning_refiner软件将两个软件生成的bin进行合并,重新生成bin,并提纯bin。
5. 去冗余和筛选:合并所有样本中得到的bins,用CheckM中的lineage_wf流程评估bins的完成度和污染度,并用dRep软件对bins进行去冗余。
6. Bin丰度计算:在metaWRAP环境中,使用metaWRAP的quant_bins模块计算每个Bin的丰度。
7. Bin物种注释:使用 PhyloPhlAn3软件将bins与SGB.Jul20比对,获取每个bin的物种分类信息。
8. Bin功能注释:基于Prokka软件得到的蛋白序列信息,用emapper软件和eggNOG数据库进行比对,得到COG、KEGG、CAZy、GO的信息,采用ARI软件和CARD数据库进行比对得到抗性基因信息,进行基因个数的统计,并进行可视化。
四、主要分析结果
1、Bin信息统计
Bin信息统计(示例)
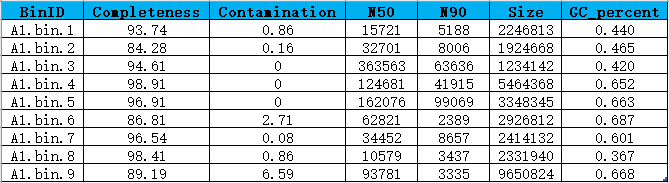
第一列:bin编号;第二列:bin完成度;第三列:bin污染度;第四列:Contig N50;第五列:Contig N90;第六列:一个Bin的所有Contig长度之和,即该基因组草图的总长度;第七列:GC含量是指一个Bin中GC碱基占总碱基的比例。
2、GC-depth图
Contig GC含量和depth深度数据即可进行可视化,绘制Bin中每个contig的散点图。此图可以用来判断分箱效果和污染情况。
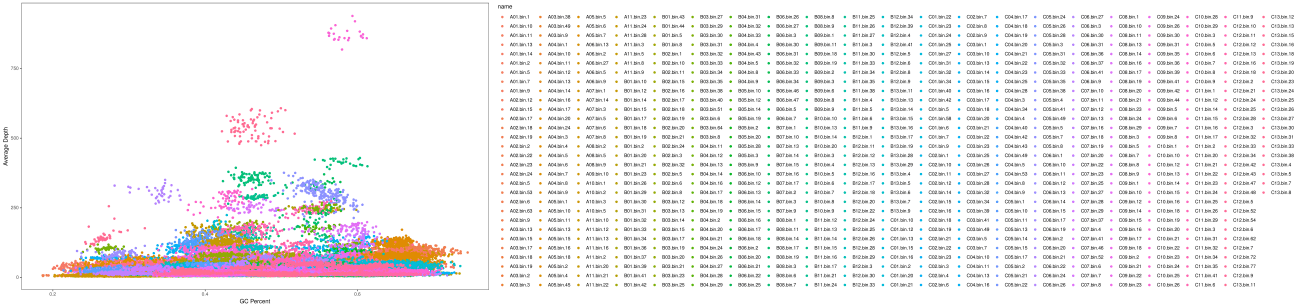 Binned contigs可视化
Binned contigs可视化
注:横坐标是contig的GC含量;纵坐标是contig depth;一个点代表一个contig,相同颜色的contig来自同一个bin。
3、GOLevel2基因数统计
GO提供了一系列的语义(terms)用于描绘基因、基因产物的特点,这些语义通过三个概念维度展开:细胞学组件(Cellular Component)用于描述某个节点的亚细胞结构、位置和大分子复合物;分子功能(molecular function),用于描述基因以及基因产物的功能;生物学途径(biological process)指的是分子功能的有序组合以实现更复杂的生物功能,
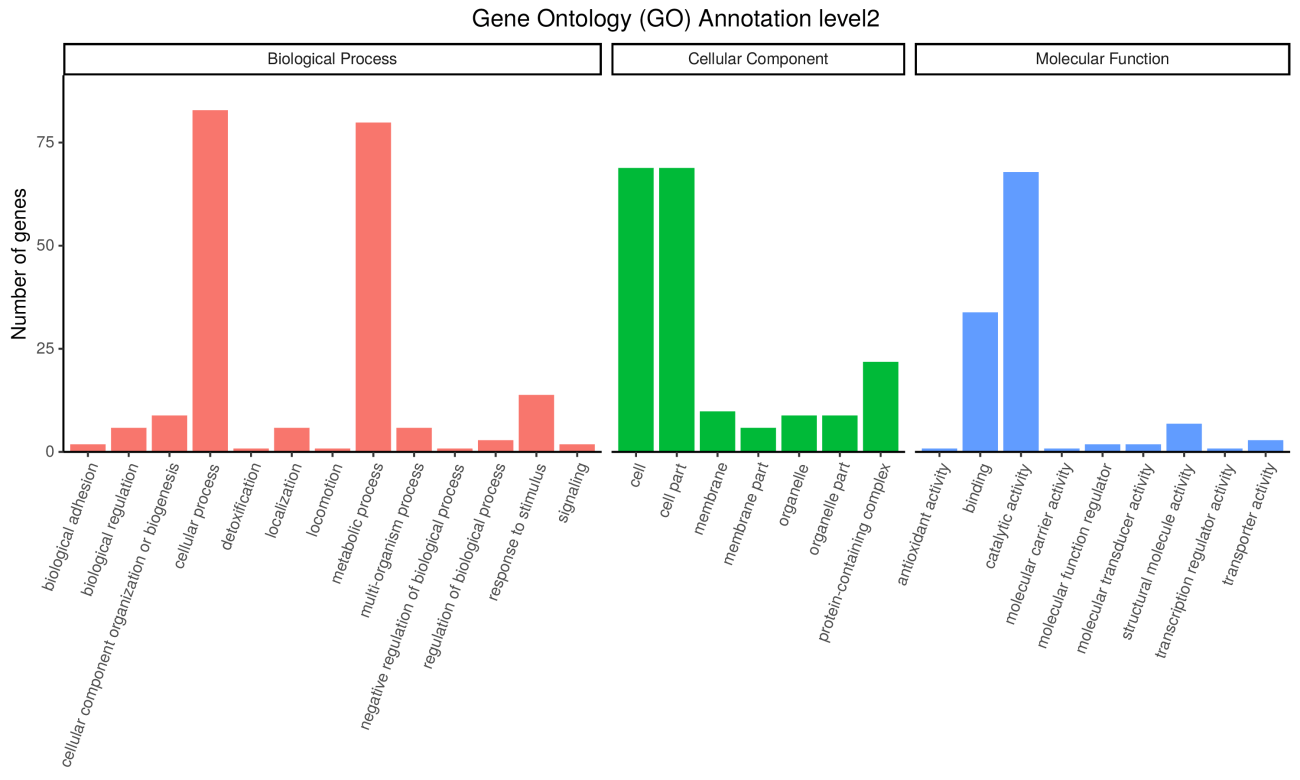 GO注释统计图
GO注释统计图
注:该图说明的是其中一个bin预测基因所属GO的情况;纵坐标是数量;横坐标是GO;颜色是GO分类;柱子越高,该GO中包含的预测基因(CDS)越多。
4、KEGGLevel2基因数统计
KEGG PATHWAY 数据库中,将生物代谢通路划分为 6 类,分别为:细胞过程(Cellular Processes)、环境信息处理(Environmental Information Processing)、遗传信息处理(Genetic Information Processing)、人类疾病(Human Diseases)、新陈代谢(Metabolism)、生物体系统(Organismal Systems),其中每类又被系统分类为二、三、四层。第二层目前包括有 57个种子 pathway;第三层即为其代谢通路图;第四层为每个代谢通路图的具体注释信息。
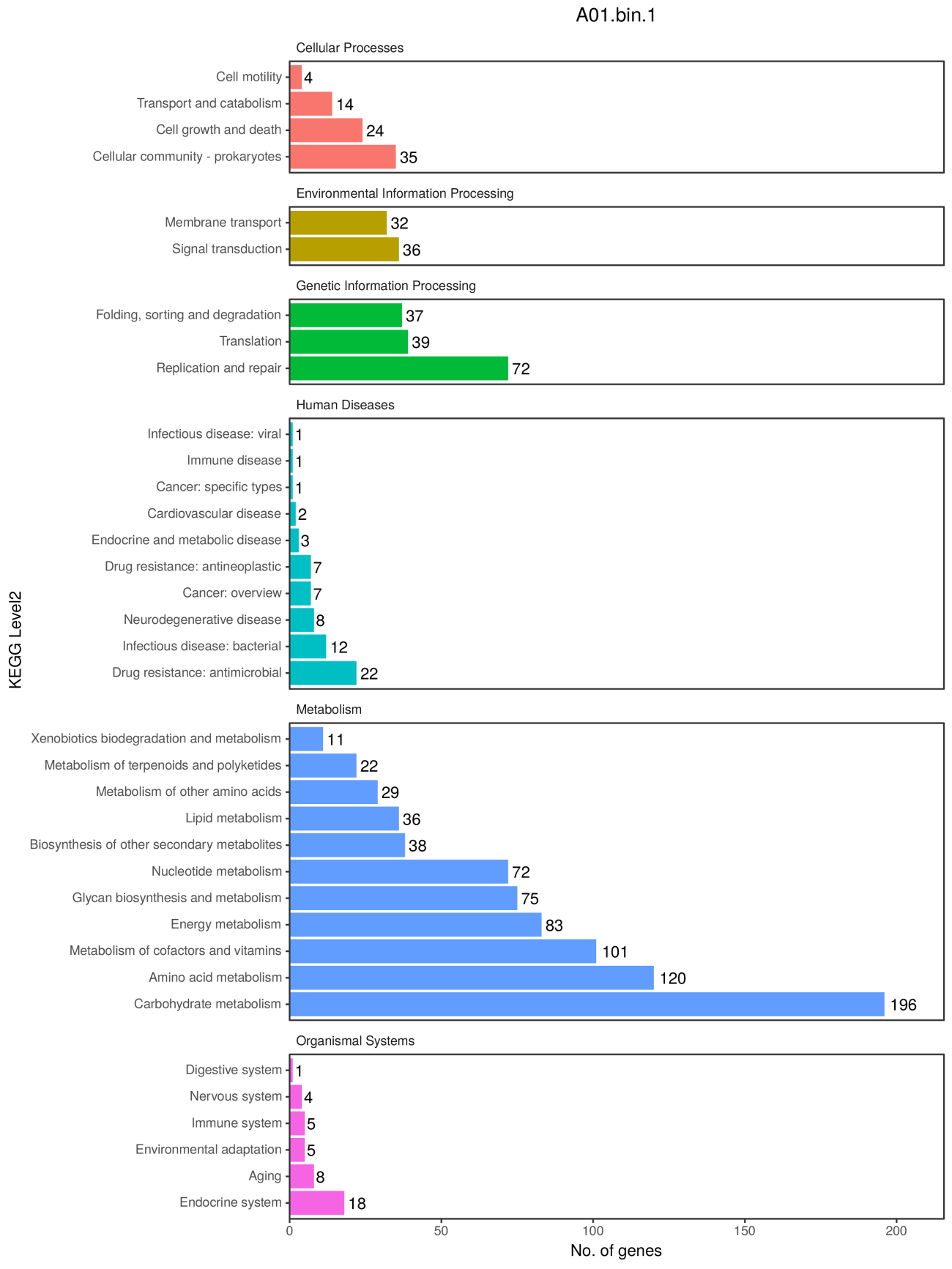 KEGG pathway level2水平CDS数量可视化
KEGG pathway level2水平CDS数量可视化
注:横坐标是KEGG level2水平CDS数量;纵坐标是KEGG level 2名称;颜色用于区分KEGG level 1的类型。
4、COG各类别基因数统计
COG,即Clusters of Orthologous Groups of proteins(同源蛋白簇)。COG是由NCBI创建并维护的蛋白数据库,根据细菌、藻类和真核生物完整基因组的编码蛋白系统进化关系分类构建而成。通过比对可以将某个蛋白序列注释到某一个COG中,每一簇COG由直系同源序列构成,从而可以推测该序列的功能。
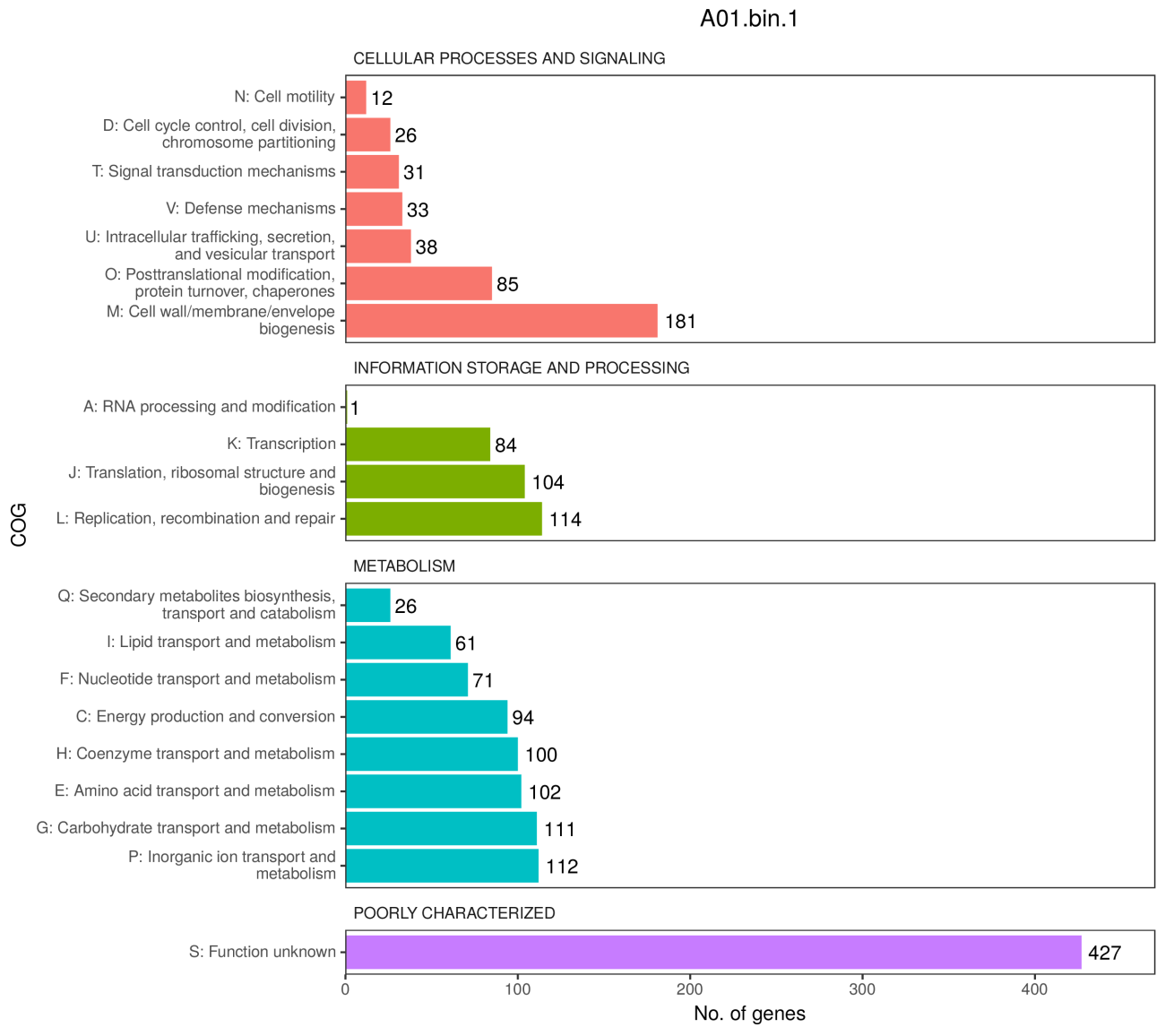
注:纵坐标是COG level 2分类(字母表示,共26种分类);横坐标是注释到的CDS计数;颜色是COG level 1分类(四大类,分别是:细胞过程和信号传递、信息储存和加工、代谢和尚未明确);柱子越长,属于该分类的预测基因越多。
4、Bin进化树
使用 PhyloPhlAn3软件将Bin与SGB.Jul20比对,以获取Bin之间的进化关系树和物种注释。图中的热图展示的是Bin相对丰度信息, 分支不同颜色表示门分类信息。

5、Bin圈图可视化
利用每个Bin(基于contig)的Prokka蛋白预测信息,功能区注释信息(含正负链、CDS、RNA类型等信息),以及GC content、GC skew的统计结果绘制Circos圈图,直观展示整个Bin的功能注释信息。
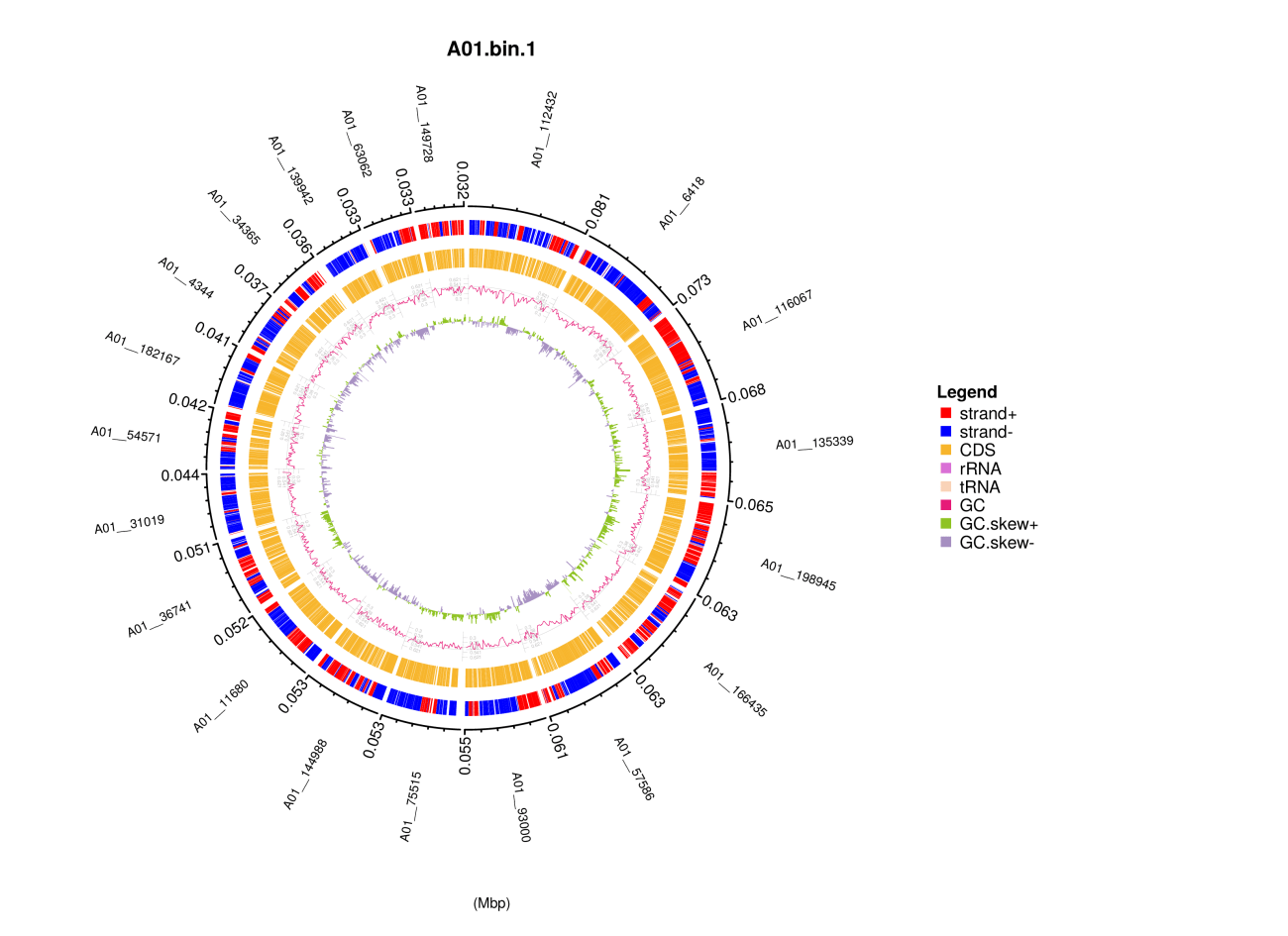
- 本文固定链接: https://maimengkong.com/zu/949.html
- 转载请注明: : 萌小白 2022年6月2日 于 卖萌控的博客 发表
- 百度已收录
