微生物重测序是基于高通量测序数据,与近缘参考基因组进行比对,进行变异检测的方法。通过重测序可以获得目标基因组对于参考基因组的SNP、InDel、SV等一系列变异信息,从中尝试对基因组之间的性状差异进行解析,或作为标记进行大规模的进化分析。本期小编将以微生物重测序分析为例给大家介绍短序列比对软件bwa的使用。
一
BWA 下载安装
-
下载https://nchc.dl.sourceforge.net/project/bio-bwa/bwa-0.7.15.tar.bz2
tar -jxvf bwa-0.7.15.tar.bz2# 解压缩
cd bwa-0.7.15
make # 编译
-
配置环境变量:
-
若要临时修改环境变量,可直接在终端输入下面一行命令:
Export PATH=/where/to/install/bin:$PATH
-
要永久修改环境变量可将下面第一行添加到~/.bash_profile(针对当前用户)或者/etc/profile(针对所有用户)文件的末尾,再执行第二行命令即可:
Export PATH=$PATH: /where/to/install/bin;
source ~/.bash_profile或者source /etc/profile
-
现在bwa已经安装好了,下面我们就利用bwa对微生物reads进行mapping
二
使用流程
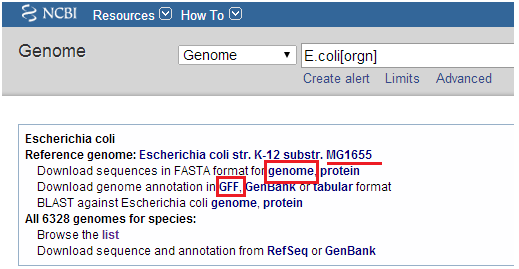
1.输入文件:我们以两个肠杆科菌株数据为例(sample1和sample2),两个菌株的测序仪下机数据fastq格式数据,和E.coli的MG1655参考基因组序列ref.fa;

Fastq文件每四行表示一个read(如上图所示),其中第一第三行表示read名称等相关信息,第二行为read序列,第四行为第二行对应的每个碱基质量值。
参考基因组文件: NCBI下载的E.coli MG1655基因组序列ref.fa和基因组注释文件ref.gff(用于变异注释)
2. bwa mapping到参考基因组
1)为参考基因组建立索引
bwa index ref.fa #参数说明:
-a BWT构建算法:bwtsw, is of rb2 [default],bwtsw适用于较长基因组,另外两个使用于短基因组;
-p 索引的前缀[same as fasta name];
-b bwtsw算法模块长度,与-a bwtsw一起使用,[default 10000000];
2)寻找SA coordinates
bwa aln ref.fa sample.fq1.gz > sample.fq1.sai # pair-end
bwa aln ref.fa sample.fq2.gz > sample.fq2.sai
bwa sample ref.fa sample.fq1.sai sample.fq2.sai sample.fq1.gz sample.fq2.gz > sample.sam
bwa aln ref.fa sample.fq.gz > sample.fq.sai # single-end
bwa samse ref.fa sample.fq.sai sample.fq.gz > sample.sam
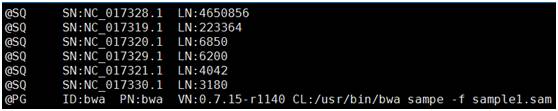
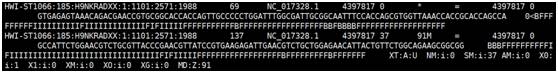
sam文件格式如下,以@开头的行为注释行,没有@开头的部分为具体比对信息,每行表示一条read与参考基因组的比对情况,每行共有12列,依次为:read name,flag,参考序列编号,比对上的位置,mapping的质量值,简要比对信息表达式,下一个片段比对上的参考序列编号,下一片段比对到参考序列上的 第一个碱基位置,参考序列和比对上的序列共同组成的序列Template的长度,序列片段信息,序列质量值信息以及可选区域(格式为TAG TYPE VALUE)。
3)将sam进行排序,并转换为bam文件
samtools sort sample.sam –output-fmt BAM –o sample.sort.bam
参数说明:
--output –fmt BAM 指定输出文件为bam格式文件;
-o 输出文件名;
统计所有位点的测序深度
samtools depth –a sample.sort.bam > sample.depth
参数说明:
-a 输出所有位点,包括深度为0的位点;
-l read长度阈值,低于该长度的read将被忽略;
-d 最大覆盖深度,默认8000;
-q 碱基质量阈值;
-Q 比对质量阈值;
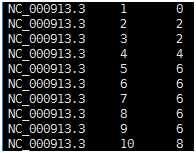
Sample.depth 文件(如下图所示)由三列组成,依次为染色体名,参考基因组位点,和该位点的覆盖深度。
Samtools 软件的安装和使用将在下期进行详细介绍。
三
参考文献
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform.[J]. Bioinformatics, 2010, 25(14):1754-1760. doi: 10.1093/bioinformatics/btp324, pubmed:19451168.
Ayat H, Doruk B, Toland A E, et al. Benchmarking short sequence mapping tools[J]. Bmc Bioinformatics, 2013, 14(1):184
- 本文固定链接: https://maimengkong.com/zu/1222.html
- 转载请注明: : 萌小白 2022年10月3日 于 卖萌控的博客 发表
- 百度已收录
