作者:微生态
编译:微科盟牛魔王,编辑:微科盟茗溪、江舜尧。
微科盟原创微文,欢迎转发转载,转载须注明来源《微生态》公众号。
导读肠道微生物群与多种生理状态有关,但关于因果关系的争议仍未解决。在本研究中,我们利用全基因组、全宏基因组、人体测量和血液代谢特征数据对3432名中国人进行了双向孟德尔随机分析。我们确定了58个肠道微生物群和血液代谢物之间的因果关系,并复制了其中的43个。粪便中Oscillibacter和Alistipes的相对丰度增加与甘油三酯浓度降低有因果关系。相反,血液代谢物如谷氨酸似乎减少了粪便中的Oxalobacter,而变形杆菌的成员则受到代谢物如5-甲基四氢叶酸、丙氨酸、谷氨酸和硒的影响。利用日本Biobank的数据进行双样本孟德尔随机化分析,部分证实了甘油三酯和尿酸的结果,也为已发表的癌症和心血管疾病的粪便细菌标志物提供了因果支持。这项研究说明了人类遗传信息的价值,有助于优先考虑肠道微生物特征的机理和临床研究。
论文ID
原名:Mendelian randomization analyses support causal relationships between blood metabolites and the gut microbiome
译名:孟德尔随机化分析揭示血液代谢物和肠道微生物群之间的因果关系
期刊:Nature Genetics
IF:38.33
发表时间:2022年1月3日
通讯作者:张涛,贾慧珏
通讯作者单位:深圳华大生命科学研究院
DOI号:10.1038/ s41588-021-00968-y
实验设计
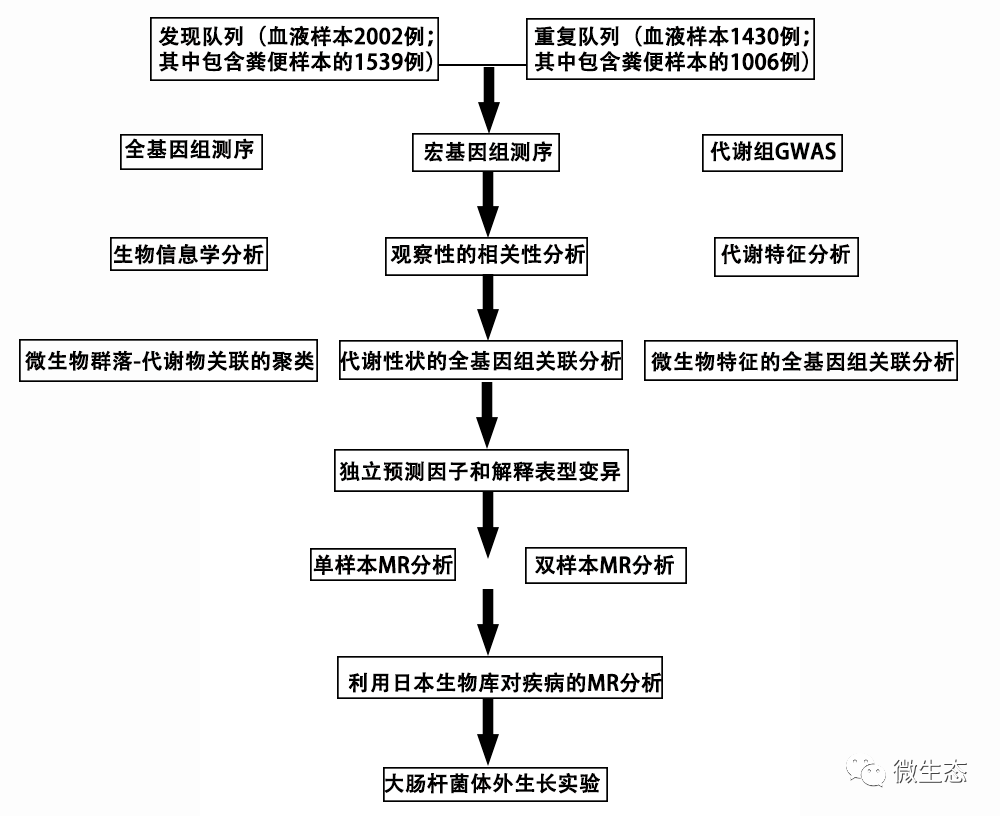
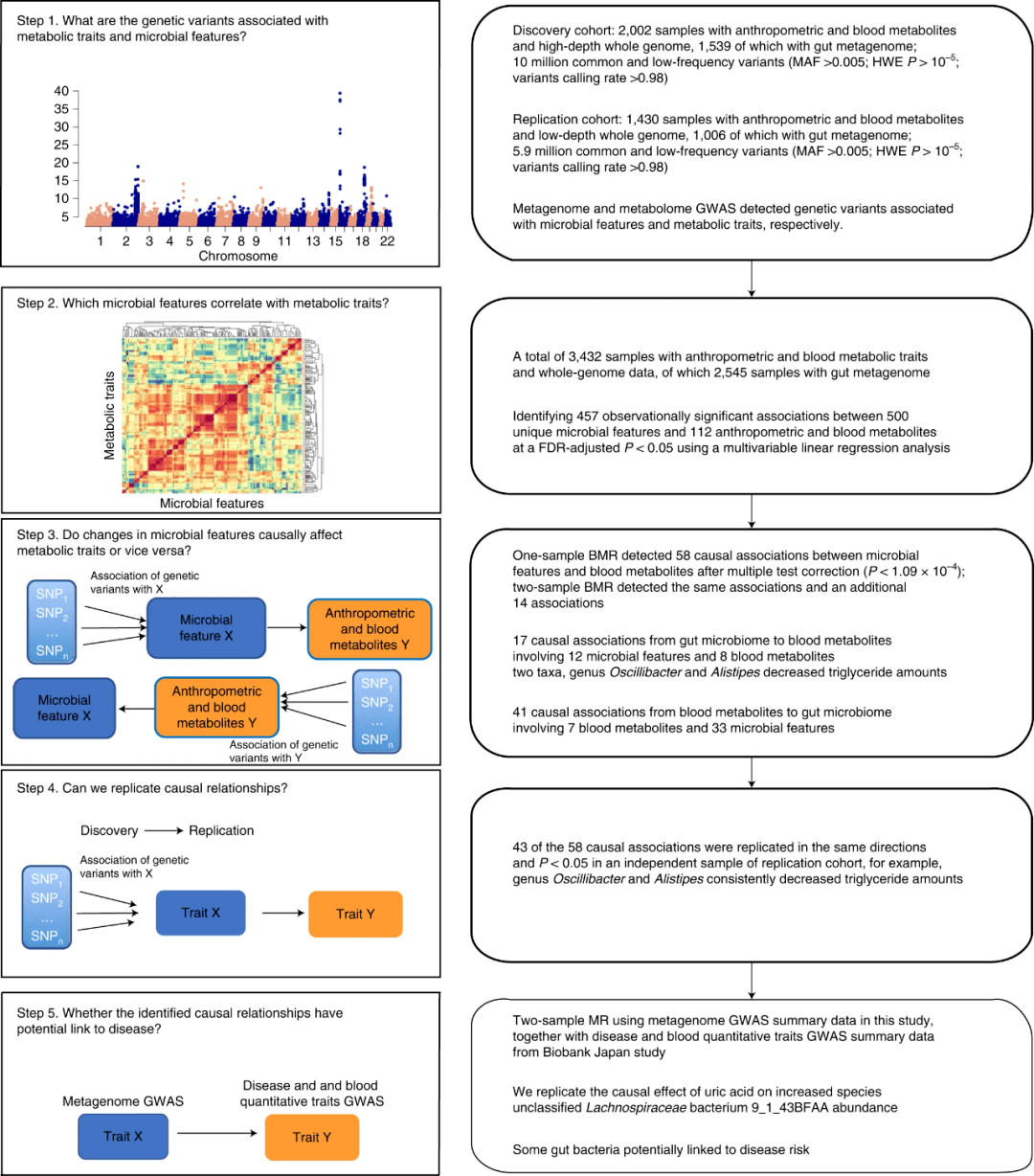
图1. 研究设计和工作流程。对于每个步骤,这个示意图突出了我们试图回答的研究问题、分析流程、使用的数据和概括的结果。我们首先进行了宏基因组和代谢组GWAS检测,分别在发现和重复队列中检测与微生物特征和代谢特征相关的遗传变异(步骤1)。然后,我们进行观察分析,以确定哪些微生物特征(类群,GMM)与该队列中的代谢特征相关(步骤2)。我们使用了2545个样本,同时包含微生物特征和代谢特征的信息。经FDR调整后,我们观察到500个独特的微生物特征与112个人体测量学和血液代谢特征之间有457个显著相关性(P<0.05)。然后,我们在发现队列中通过BMR分析评估了457个观察性关联的因果关系(步骤3)。经多项检测校正后,单样本基础磁共振检测出58个微生物特征与血液代谢物之间的因果关系(P<1.0 9×10−4);2个样本基础磁共振检测到相同的关联,另外14个关联。作为验证,我们在独立的复制队列中使用相同的MR分析复制了已发现的因果关系(步骤4)。在58个因果关系中,超过一半(43个)是在同一方向上复制的(P<0.05)。最后,我们使用2个样本的磁共振分析来调查从Biobank Japan研究(步骤5)确定的72个因果关系对疾病的影响。
结果
1 与人类基因变异相关的粪便微生物群特征
我们着手确定人类遗传变异作为MR的随机层(图1)。4D-SZ发现队列包含了来自2002份血液样本的高深度全基因组测序数据(平均深度42x;附表1,附图1a),其中1539人拥有从粪便样本中获得的宏基因组鸟枪测序数据(8.56±2.28 Gb;附图1b)。我们利用了1000万个常见和低频变异体(次要等位基因频率(MAF) ≥0.5%)和500个独特的微生物特征(Spearman’s相关性<0.99)。M-GWAS根据年龄、性别、体重指数(BMI)、排便频率、粪便形态、自我报告的饮食、生活方式因素和基因组数据中的前四个主要成分(PCs)进行调整,以解释人群分层。
在全基因组和宏基因组数据的大队列中,我们进行了M-GWAS分析,并确定了总共625个关联,涉及548个独立的基因座,对500个微生物特征中的1个或多个在基因组上有显著性(P<5×10−8)。在整个范围内,采用更保守的Bonferroni校正P值差异显著,为1.0× 10−10 (=5×10−8/500)。本文确定了28个与粪便微生物特征相关的基因位点,涉及27个基因组位点,其中5个与肠道细菌相关,其余22个与肠道代谢途径相关(附表2)。
对于MR来说,所用的遗传变异能代表微生物组的特征是很重要的,因此,像以前的MR研究一样(附图2),使用了小于1×10-5的提示性的P值(附表2)。每个微生物特征平均有44个遗传突变(图2a;附表3)。相应的遗传突变对微生物特征的解释中位数为24.9%,例如,消耗琥珀酸的微生物代谢途径为45.5%,Phascolarctobacterium succinatutens(一种无糖的、利用琥珀酸的细菌)为44.6%,而Edwardsiella属只有6.8%(附表3)。5个属 (Bilophila、Oscillibacter、Faecalibacterium、Megasphaera和Bacteroides) 的表型(相对丰度)差异可由其相应的独立遗传变异解释35%以上(图2b)。因此,尽管人类遗传关联(阵列数据)被报道只解释了10%或1.9%的肠道微生物群,但从目前的M-GWAS研究中推测的关联可以高度预测某些肠道类群和功能。
为了更好地确定这些相关性,我们对来自中国多个城市的1430个个体的重复队列进行了测序(其中对粪便样本也进行了宏基因组测序,但人类基因组采用大约8倍的全基因组测序;附图1c,d)。在发现队列中确定的22293个独立关联中(P <10-5),有4,876个突变体在低深度重复数据集中不可用,其中87.6%的变体不是常见突变体(MAF<0.05)。在剩余的17417个独立关联中,我们能够复制出的2324个小等位基因相同效应方向(P<0.05;附表2),表明这些关联不是随机的假阳性。使用P<10-5的阈值(2,324/17,417)在同一方向上复制的关联比例(P<0.05)并不低于更严格的阈值(54/625的P<5×10-8,和2/28的P<10-10)。在整个研究的阈值中,2个重复性好的信号是ABO血型中的rs1461780285,与模块MF0007有关:乳糖和半乳糖降解(Pdiscovery =2.10 × 10-12,Preplication = 1.09 × 10-10;附图3a,b)和LCORL基因附近的rs142693490(牵涉到精子生成、体格和身高),与MF0034有关:丙氨酸降解II(Pdiscovery = 1.28 × 10-12,Preplication = 0.014;附图3c,d)。遗传变体rs1461780285与ABO基因中的多个单核苷酸多态性(SNPs)(rs507666、rs532436、rs651007、rs579459和rs579459)处于强连锁不平衡状态(LD,r2=0.99)。在本研究和以前的研究中,发现这些位于一个区块的SNPs与代谢物水平有关,尤其是血清碱性磷酸酶水平(附表4)。此外,我们能够重复先前报道的几个微生物信号,特别是rs12354611和Bacteroides stercoris(P = 8.64 × 10-6;附表5和补充说明)。
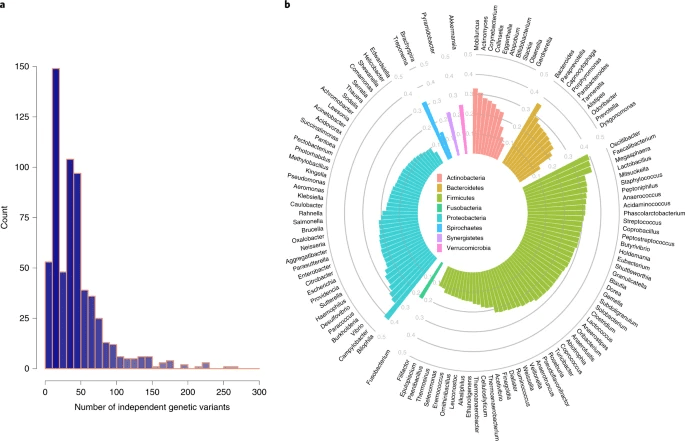
图2. 微生物特征的独立遗传变异及其解释的变异。a.密度图,显示了与每个微生物特征相关的独立遗传变异的数量分布(r2<0.1和P<1×10−5),通过GWAS分析计算出总共5 0 0个独特的微生物特征。X轴表示每个微生物特征(分类单元或GMM)的独立遗传变异体的数量。Y轴表示给定数量的独立预测因子下微生物特征的数量。b.方差由每个微生物特征对应的独立遗传变异解释。柱状极坐标图显示了每个常见属的独立遗传变异(出现在至少50%的样本中)在很大程度上解释了它们的表型差异(每个属的相对丰度)。属根据各自的门进行分类,并用不同的颜色标记。在GCTA工具中使用REML方法计算h2。
2 与人类基因变异相关的血液代谢特征
另一方面,众所周知血浆代谢物水平与宿主遗传变异相关(图1)。因此,我们对112种代谢物中的每一种进行了全基因组的关联测试,并对相对浓度进行了对数转换。我们共发现了174个关联,涉及158个基因座,这些基因座与112种代谢物中的1种或多种有全基因组意义(P<5×10-8)的代谢物有独立关联。采用更保守的Bonferroni校正的全研究范围的显著P值,为4.5×10-10(=5×10-8/112种代谢物),我们确定了涉及28个基因组位点的39个与代谢物相关的基因(附表6)。这些关联包括先前已确立的关联,如UGT1A家族与血清总胆红素相关,ASPG与天冬酰胺相关。
根据P<10-5的提示性阈值,我们确定了6541个代谢定量性状位点,其中361个与2个或更多的代谢物相关(附表6)。每个代谢性状的平均遗传变异数为58(图3a;附表7)。相应的遗传变异所解释的变异百分比从13.3%(红血球分布)到高达48.3%(血汞浓度)和45.9%(血甲胎蛋白值),中位数为28.6%。其中,2268个变异体或其代理变异体(r2>0.6;距离<1Mb)已被GWAS目录报道(附表8)。我们还发现,一些变异与GWAS目录中的疾病相关,如慢性肾脏病、阿尔茨海默氏病、冠状动脉疾病、克罗恩病、卵巢癌、乳腺癌和胃癌。
在4D-SZ发现队列P<10−5的6,541个提示性代谢数量性状基因座中,有5,088个变异被重复数据集覆盖,在相同的小等位基因效应方向上重复,其中717个和31个分别在名义 (P<0.05) 和建议上有显著差异 (P<10−5)。特别是对于174个全基因组显著关联和39个全研究显著关联,我们可以在同一方向上分别重复51个和29个关联 (P<0.05)。低深度基因组 (在发现和重复队列中P<4.5×10−10) 确认的前几个关联包括:与锰相关的FECH;与血清总胆红素以及直接和间接 (未结合) 胆红素相关的UGT1A家族;与天冬酰胺相关的天冬氨酸转氨酶;与甘氨酸相关的CPS1;以及与低密度脂蛋白相关的载脂蛋白(附图4)。总体而言,准确识别遗传决定因素,以及微生物特征和血液代谢物的高方差解释是磁共振分析调查因果关系的最佳选择。
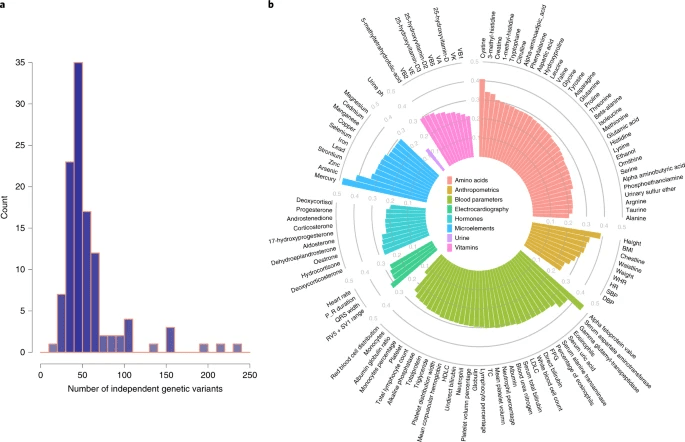
图3. 代谢性状的独立遗传变异及其解释的变异。a.密度图,显示与每个代谢性状相关的独立遗传变异数的分布(r2<0.1和P<1×10−5)。X轴表示每个代谢性状的独立遗传变异的数量。Y轴表示在给定数量的独立预测因子下的代谢特性的数量。b. 方差由每个代谢性状对应的独立遗传变量解释。极柱图显示了每个代谢性状的独立遗传变异在多大程度上解释了它们的表型变异。每个代谢性状被归入不同的类别,并用不同的颜色标记。用GCTA工具中的REML方法计算h2。
3 从观察相关性到MR
作为强因果关系的前提条件,我们用多元线性回归方法研究了500个独特的粪便微生物特征(类群和功能模块)与112个寄主代谢性状的相对丰度之间的相关性。在调整性别和年龄后,我们观察到457个有意义的相关性(错误发现率(FDR)校正P<0.05;附表9)。3种代谢产物谷氨酸、5-甲基四氢叶酸(5-甲基四氢叶酸,叶酸的活性形式)和硒与大量的微生物特征有关(补充图5)。这些关联扩展了多项研究成果,并提出肠道微生物类群/功能与血浆代谢物之间的定量关系。
由于我们幸运地拥有同一队列中的所有数据,我们首先进行了单样本MR分析,以确定发现队列中457个观察相关性的因果关系,该队列由1539个同时具有代谢和微生物组特征的个人组成。我们发现58个显著的因果效应,其中17个是肠道微生物性状对血液代谢性状的因果效应,另外41个是血液代谢性状对肠道微生物性状的因果效应(P<1.09×10=0.05/457;图4;附表10)。其中只有4个是双向的。通过对来自不同城市的1006个低深度基因组以及代谢和微生物组特征的复制数据集进行单样本MR分析,我们可以复制58个因果关系中的43个(方向相同,P<0.05;附表10),表明这些影响不是随机的假阳性。
此外,我们还使用了6种不同的双样本磁共振方法(当只能从2个不同的队列获得汇总统计数据时更常用)来分析我们在发现队列(2,002个具有代谢特征的样本和1,539个具有微生物特征的样本的摘要数据)和重复队列(1,430个具有代谢特征的样本和1,006个具有微生物特征的样本的摘要数据)中的数据。单样本MR和双样本MR分析结果高度一致,发现队列中单样本和双样本MRβ系数的Spearman相关系数达到0.767(P<2.2×10−16)。单样本MR确定的58个因果关系在双样本MR分析中也很重要。本文还通过2个样本的MR分析确定了另外14个因果关系(附表11),可能是由于队列规模较大。此外,我们还使用MR-PRESO全球测试检查了水平多效性的存在。只有1个因果关系(硒对甲基鞭毛菌丰度的负面影响,PMR-PRESSOGlobaltest=0.01;附表9)表现出多效性,而其余71个因果关系没有表现出多效性(P>0.05)。因此,我们的MR分析确定了血液代谢特征和肠道微生物群的特定特征之间的强大因果关系。
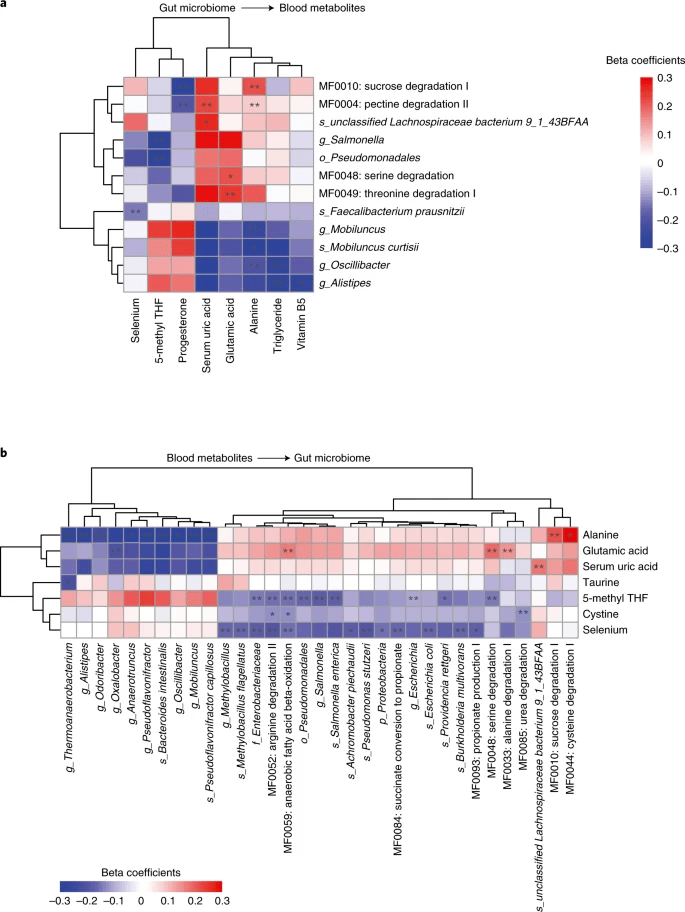
图4. 确定微生物特征和代谢特征的58个因果关系。a图显示了12个特定微生物特征对8个代谢性状的因果影响,这8个代谢性状涉及从肠道微生物群到血液代谢物的17个因果关系。b图显示7种血液代谢物对33个微生物特征的因果影响的曲线图,涉及从血液代谢物到肠道微生物群的41个因果关系。用双星号标记的细胞代表在发现队列中识别的58个关联中的43个,这些关联也在复制队列中复制,而单个星号代表仅在发现队列中有意义的其他15个关联。细胞根据一个样本MR分析的β系数进行着色,红色和蓝色分别对应于正相关和负相关。
4 肠道微生物群对血液代谢特性的影响
由于某些MR鉴定的关系出现关联,我们对从肠道微生物群到血液代谢物中涉及到17种因果关系的12个微生物特征和8个血液代谢物进行了分层聚类,并形成了2个聚类。一组通过肠道微生物类群或功能模块降低血浆甘油三酯和丙氨酸水平,另一组包括通过肠道微生物特征降低5-甲基四氢呋喃或孕酮水平,但增加血清尿酸或血浆谷氨酸水平(图4a)。令人欣慰的是,Curtisii Mobiluncus与其对应的Mobiluncus属聚在一起,其模块包括丝氨酸降解和苏氨酸降解、蔗糖降解和果胶降解。
最显著的因果作用是Oscillibacter降低血甘油三酯浓度(图5a-c), 并在较小程度上降低BMI和腰臀比(WHR),而血浆丙氨酸的影响是双向的。我们用134个遗传变异构建多基因风险得分(PRS)分别解释39.3%和49.6%的表型变异(图3b;附表10)。对于发现队列中的单样本MR分析,我们估计, Oscillibacter 丰度每增加1s.d.,甘油三酯浓度会下降0.261 mmol l −1 ( P =2.53×10 −10 ),体重指数下降0.161 kg m −2 ( P =1.33×10 −4 ),腰臀比下降0.126( P =2.73×10 −3 )。当进行4个双样本MR ( PGCTA-GSMR (genome-wide complex trait analysis-generalized summary MR)=4.34×10−11,PInverse_Variance_Weight=2.45×10−15,Pweight-Medium=1.22×10−7和Pmr-Egger=1.35×10−5) (图5C)时,这种因果关系是可靠的,并且没有水平多效性的证据 ( PMR-PRESSOGLOBALTEST =0.18)(图5C; 附表11);反向MR分析(检验甘油三酯的遗传预测因子对Oscillibacter丰度的影响)是显著的,但未达到多重检验校正的显著性(10−4这些细菌对于具有合适遗传背景的个体是很有希望的补充剂。
肠道微生物群降解果胶II的潜力(42.6%的变异可以用遗传风险评分来解释)显示出几个与血液性状相关的显著的MR结果(图4a),包括对丙氨酸(P=8.57×10−5)和血清尿酸(P=1.34×10−6)的正面影响,对孕酮(P=6.68×10−7)的负面影响。Bacteroidetes和 Fusobacteria是仅有的两个与果胶降解II的丰度呈正相关的门(Spearman’s秩相关,ρ分别为0.48和0.15),其中包括先前报道的2种降解果胶的Bacteroides thetaiotaomicron和Fusobacterium varium。在4D-SZ队列中,F. varium与果胶降解II(Spearman’s相关性,ρ=0.12),血丙氨酸(P=0.02)和血尿酸(P=0.04)升高相关;而B. thetaiotaomicron与果胶降解II相关(Spearman’s相关性,ρ=0.21),但对丙氨酸和尿酸没有明显影响(P>0.05;附图7a,b和c)。相反,与果胶降解II(Spearman’s相关性,ρ=0.32;附图7c)相关性最强的细菌Bacteroides Dorei,丙氨酸(P=0.05)和血清尿酸水平(P=3.40×10−4;附图7d)正相关。
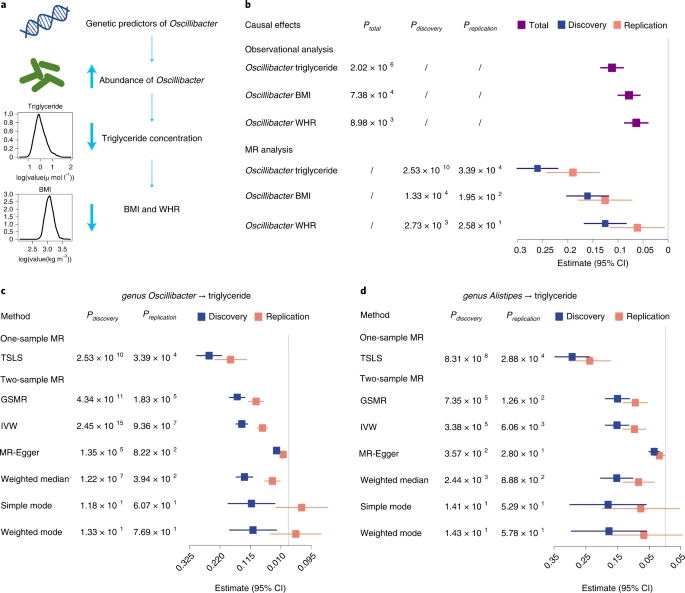
图5. Oscillibacter和Alistipes 降低血甘油三酯浓度的因果效应。a.MR分析结果示意图:具有较高丰度的Oscillibacter的遗传易感性与降低血液甘油三酯浓度有关,在较小程度上与BMI和WHR的降低有关。b森林图表示每增加1秒Oscillibacter丰度对血液甘油三酯、BMI和WHR的影响,分别使用观察和MR分析进行估计。使用多变量线性模型对总共2545个样本(紫色)进行观测相关分析。通过使用由多达134个遗传预测因子构建的PRS作为工具变量,分别在发现队列(蓝色)和复制队列(红色)中进行了单样本MR分析。文中列出了来自观察性和单样本MR分析的β估计和95%可信区间(CI)值以及P值。c,d.森林图分别表示Oscillibacter(c)和Alistipes(d)对甘油三酯数量的MR估计值和95%的CI值,在发现(蓝色)和重复(红色)队列中,分别使用一个样本MR和六个不同的双样本MR方法进行估计。文中还列出了每种MR方法计算的P值。
5 血液代谢产物对肠道微生物特性的影响
对于从血液代谢特征到肠道微生物特征的41个因果关系(一个样本MR;附表10),分层聚类显示了两种聚类,一种主要涉及血浆丙氨酸或谷氨酸降低细菌丰度,另一种涉及硒或5-甲基四氢呋喃(图4b)降低细菌丰度。Faecalibacterium prausnitzii对血浆硒有负面影响(图4a),而血浆硒对肠道变形杆菌如肠杆菌科(如Escherichia coli,P=3.79×10−5)、Pseudomonas stutzeri (P=1.06×10−6)以及精氨酸降解II(P=2.65×10−6)、琥珀酸转化为丙酸(P=3.55×10−5)和厌氧脂肪酸β氧化(P=9.71×10−5)等模块有负面影响。
变形杆菌门的细菌不仅受到硒的负面影响,而且受到5-甲基四氢呋喃(图4b)的负面影响。我们在体外直接验证了5-甲基四氢呋喃对大肠杆菌的作用。与低浓度或不添加5-甲基四氢呋喃相比,添加5-甲基四氢呋喃确实减缓了大肠杆菌菌株AM17-9的生长(附图8)。
少数细菌也受到谷氨酸的影响。谷氨酸对Oxalobacter (P=1.56×10−6)的负面影响(48个具有暗示关联的变异体和构建的PRS分别解释了表型变异的24.9%和2 5.4%),可能有助于解释发达国家Oxalobacter发病率较低,以及草酸摄入量和抗生素使用量较低的原因。限制谷氨酸是否能增加Oxalobacter和预防肾结石还有待检验。谷氨酸对蜂蜜二糖的降解(2个样品MR为葡萄糖、半乳糖,P=2.05×10−5)呈负向影响,对丙氨酸降解Ⅰ(P=5.46×10−5)、厌氧脂肪酸β氧化(P=9.36×10−5)和丝氨酸双向降解(分别为P=6.85×10−7和P=9.90×10−6)表现出正面影响。
6 肠道微生物群与疾病之间的因果关系
考虑到日本人的遗传结构与中国人相似,我们进一步研究了涉及40个微生物特征和12个代谢性状的72个显著因果关系(附表11)对疾病的影响,通过对4D-SZ队列的肠道微生物GWAS汇总数据和血液定量性状进行两份样本MR分析(图1;附表12)。日本生物库的研究只包括常规血液参数,而不包括氨基酸、激素或微量元素。因此,在涉及甘油三酯和血清尿酸的72个因果关系中,只有5个可以在日本生物库中进行进一步研究。在4D-SZ队列中,未分类的Lachnospiraceae bacterium 9_1_43BFAA与尿酸的关系是相互的,我们可以重复尿酸对日本队列中未分类的Lachnospiraceae bacterium 9_1_43BFAA丰度增加的因果效应,而相互作用,即未分类的Lachnospiraceae bacterium 9_1_43BFAA对尿酸的潜在影响没有被复制,这可能是因为在日本的基因型队列中缺乏变异(15个而不是67个;附表13)。另外,其他三个关联没有被复制,可能是出于同样的原因。例如,在我们汇总的数据中,Oscillibacter属有135个变异体,P<10−5,但在日本生物库数据中只有15个变异体。
使用我们的肠道微生物组M-GWAS汇总数据和日本GWAS汇总统计数据进行MR推断发现,在4D-SZ队列中,Alistipes对血液甘油三酯有负面影响,降低了脑动脉瘤(附表14;P=4.61×10−4)和肝细胞癌(P=0.045)的风险。根据我们确定的变形杆菌的遗传关联,我们能够在日本生物库疾病数据中看到变形杆菌纲增加了T2D(图6A;P=7.61×10−4,双样本MR)、充血性心力衰竭(P=0.003)和结直肠癌(P=0.047)的风险。这与主要针对肠杆菌科的MWAS研究结果一致,并表明上述代谢物(5-甲基四氢呋喃,硒)可能有助于预防这些疾病。叶酸确实被推荐用于心脏病。此外,大肠杆菌增加了尿石症(图6b;P=0.009)和肝细胞癌(P=0.04)的风险,但降低了间质性肺病的风险(P=0.007)。同样,Salmonella enterica增加了前列腺癌风险,但降低了间质性肺部疾病的风险。Pseudomonadales是唯一对肺结核有积极作用的微生物特征。异位性皮炎(P=0.005)、胃癌(P=0.008)、食道癌(P=0.027)和胆道癌(P=0.034)的风险增加。据报道,在动脉粥样硬化性心血管疾病患者中相对缺乏的Bacteroides Enterinalis,但随着钾的增加而增加,而B. intestinalis对癫痫有负面影响。Streptococcus parasanguinis 对结直肠癌和后壁厚度(超声心动图)有正面影响,与MWAS研究一致。这些结果说明了肠道微生物-血液代谢物关系在理解和预防心脏代谢性疾病和癌症方面的潜在意义。
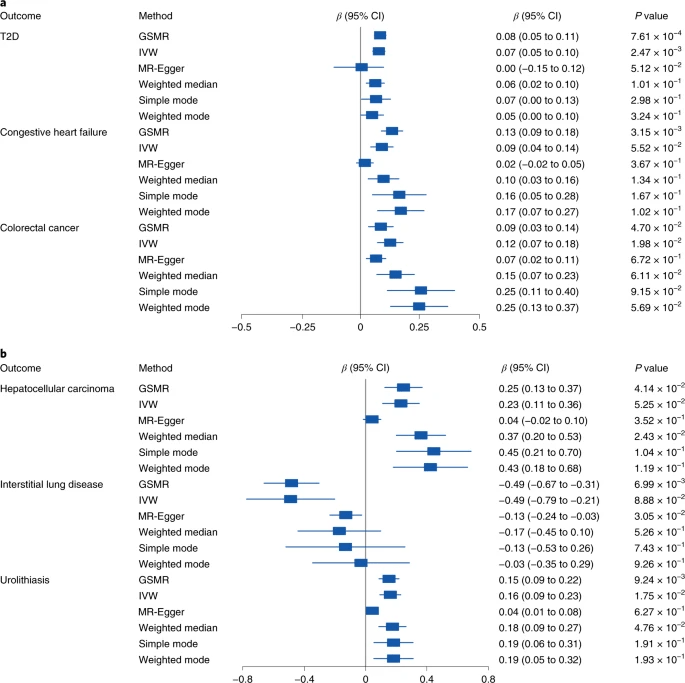
图6. Proteobacteria 和 Escherichia coli对疾病的因果作用。a.b.森林图表示Proteobacteria(a)和Escherichia coli(b)对疾病的因果效应的MR估计值和95%CI值,使用6种不同的双样本MR方法(GSMR、IVW、MR-Egger、加权中位数、简单模式和加权模式)进行估计。我们显示了估计的WGS值(95%CI),我们使用来自日本Biobank的疾病汇总统计数据和来自这个发现队列的肠道微生物组的GWAS汇总数据,显示了由每种MR方法估计的β值(95%CI)和P值。
讨论
我们发现了许多与微生物特征和代谢特征相关的遗传位点,并通过单样本BMR发现了肠道微生物群和血液代谢物之间的58个因果关系。在58个单样本MR信号中,43个可以在同样来自中国的低深度基因组测序队列中复制。双样本MR在同一方向上重复了58个因果关系,并确定了另外14个因果关系。通过两个样本MR分析来自日本Biobank的汇总数据发现了肠道微生物特征对疾病的影响,暗示了微生物组干预在心脏代谢、肾脏和肺部疾病以及癌症方面的潜在应用。虽然使用无菌小鼠和参考细菌菌株的机制研究很流行,但我们的数据驱动分析强调了尚未被培养和广泛表征的肠道微生物(例如Oscilibacter和Alistipe)的临床相关性,并降低甘油三酯浓度和许多疾病风险,这可能与东亚地区的生活方式和疾病特征快速变化非常相关。
虽然肠道微生物群和血液特征(如氨基酸和维生素)之间的联系已经被报道有一段时间了,但我们的MR分析可能会激发更多的机制和干预性研究。从4D-SZ队列中获得的独特数据可以对硒等被忽视的特征进行评估。有趣的是,F. prausnitzii对血浆硒有不良影响,同时硒对γ-变形菌产生相反的影响。氮是许多生态系统的有限资源。在现代人类肠道微生物群中,在没有高亚硝酸盐摄入的情况下,蛋白质可能是氮的主要来源,而谷氨酸-谷氨酰胺库是过量胺炎症潜在的关键缓冲机制。在这些中国人(平均年龄不超过30岁)中观察到的Proteobacteria的增加和Oxalobacteraceae的减少可能是晚年心脏代谢和肾脏疾病易感性的潜在原因。锶和Streptococcus parasanguinis之间的双向联系意味着水源和心血管疾病之间的相互作用。
不能被宿主直接消化的多糖代谢是肠道微生物群的一个重要功能。我们发现果胶(或蔗糖)的降解会对孕酮水平产生负面影响。这是一个有趣的可能性,可以为孕妇的传统饮食建议提供科学支持,以确保足月妊娠。高尿酸血症和痛风在东亚是一种日益严重的流行病,而含有果糖的软饮料是一个强力因素,其重要性不亚于啤酒和肉类。肠道微生物(拟杆菌、梭杆菌)果胶降解模块对循环丙氨酸和尿酸水平有正面影响。为了对尿酸和丙氨酸水平进行个性化管理,需要进一步研究碳和氮的跨界代谢通量。
对于M-GWAS和微生物组MR这两个新兴领域,统计专家也有很多方法论发展的机会。低频微生物在人的肠道中很常见,可能扮演生理或病理角色。我们对肠道微生物物种的MR结果与更高分类单位如属或门的MR研究结果一致(图4和附表11)。在较大的类群中,P值有时更为显著,表明其他物种也有类似的功能。自从微生物组领域研究开始,微生物组中的功能冗余就一直被讨论,在本文中,我们确定了整个研究范围内宿主与肠道微生物功能模块的显著遗传关联,以及其他肠道微生物功能模块对宿主循环代谢物水平的因果影响。除了人类等位基因频率之外,微生物组分类或功能数据的分布构成了另一层考虑因素。收集更同质的队列可以在相对较小的队列中识别信号,而对不同群体进行比较的校正可能涉及宿主-微生物群的相互作用。由于肠道微生物群可以受到药物的影响,而且大多数性状的遗传率在年轻个体中更高,健康的年轻人可能更适合作为M-GWAS的研究对象,而老年人的微生物群与药物的相互作用可能是MR研究的一个重要方向。
总体而言,我们的数据强调了M-GWAS和MR在全面了解微生物群方面的巨大潜力。这可以从机制上说明问题,并有助于聚焦干预措施,以减轻炎症和预防或缓解复杂疾病。
- 本文固定链接: https://maimengkong.com/zixun/1429.html
- 转载请注明: : 萌小白 2023年4月14日 于 卖萌控的博客 发表
- 百度已收录
