宏基因组组装有两种常见策略,一种是基于序列overlap关系进行拼接,代表软件有Omega;另一种就是基于de Bruijn图进行组装。
基于序列overlap策略组装所要求的序列长度都比较长,因此这种策略更有利于跨越短重复序列,降低宏基因组中复杂的重复序列因素带来的干扰,参见上一篇文章《为什么宏基因组组装比一般的组装困难?》。
由于现阶段的主流测序方法是二代短片段测序,序列短而且数目庞大,如果利用overlap关系直接进行组装,这要求每对reads之间都进行一次序列比较,这会很耗费时间,而且结果并不可靠。为迎合二代测序的特点,一种基于k-mer的de Bruijn组装策略则成为更有效的解决方法。
这种方法是通过把reads打断成一定长度的k-mer,然后根据k-mer之间严格的碱基配对关系构建de Bruijn图,最后通过对图形的解读找出最合理的序组装结果。
由于相邻k-mer之间的非配对碱基只有1个,那理论上相邻序列的组合情况只有四种,这大大减少了相邻序列配对的检测难度,因此de Bruijn方法是现阶段的主流组装方法。
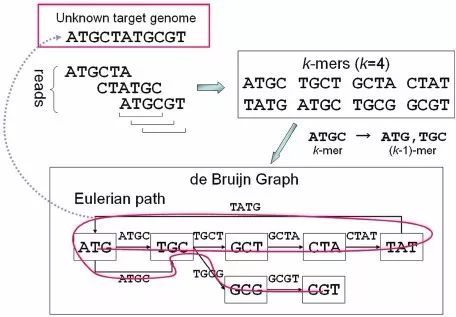
de Bruijn组装原理
k-mer 是如何影响组装效果呢?
既然de Bruijn组装策略是基于k-mer,那k-mer的深度和长度是否对测序造成影响?答案是肯定的。深度的影响我们暂且不讨论,这涉及到覆盖度和序列矫正等问题。那么k-mer的长度是如何影响组装的效果呢?其实简单来说,就是主要影响组装的特异性与敏感性。
我们看下图,假设只有两天测序序列,分别为图a中的序列1(ATCAAGCG)和序列2(CGTAAGTAC)。如果将它们打断成长度为5的k-mer,那么无论序列1还是2所产成的kmer频数都是1,经过拼接之后,都能独立组装出唯一的一条序列。也就是说,长k-mer(一般为reads长度的60%)能够有效保证k-mer之间的序列非重叠性,从组间结果更为简单。
同时,我们也可以看到,其实reads1末端和reads2前端是有CG两个碱基重叠的,也就是reads1和reads2很有可能是一条相邻的序列。但由于相邻k-mer之间必须要有k-1个碱基配对才能被连接起来,而当k-mer长度为5的时候,需要4个碱基的配对,read1和read2这种只有两个碱基配对的,明显不能被拼接起来,因此,最终k-mer=5这种策略,只能拼接成两个独立的片度。
但如果通过图b这种策略,把k-mer长度设定为2,那么,相邻k-mer只需要一个碱基的配对就能被连接,因此,read1和read2就可以通过末端2个碱基的配对被拼接起来,提供reads的利用率和reads之间拼接的敏感性。
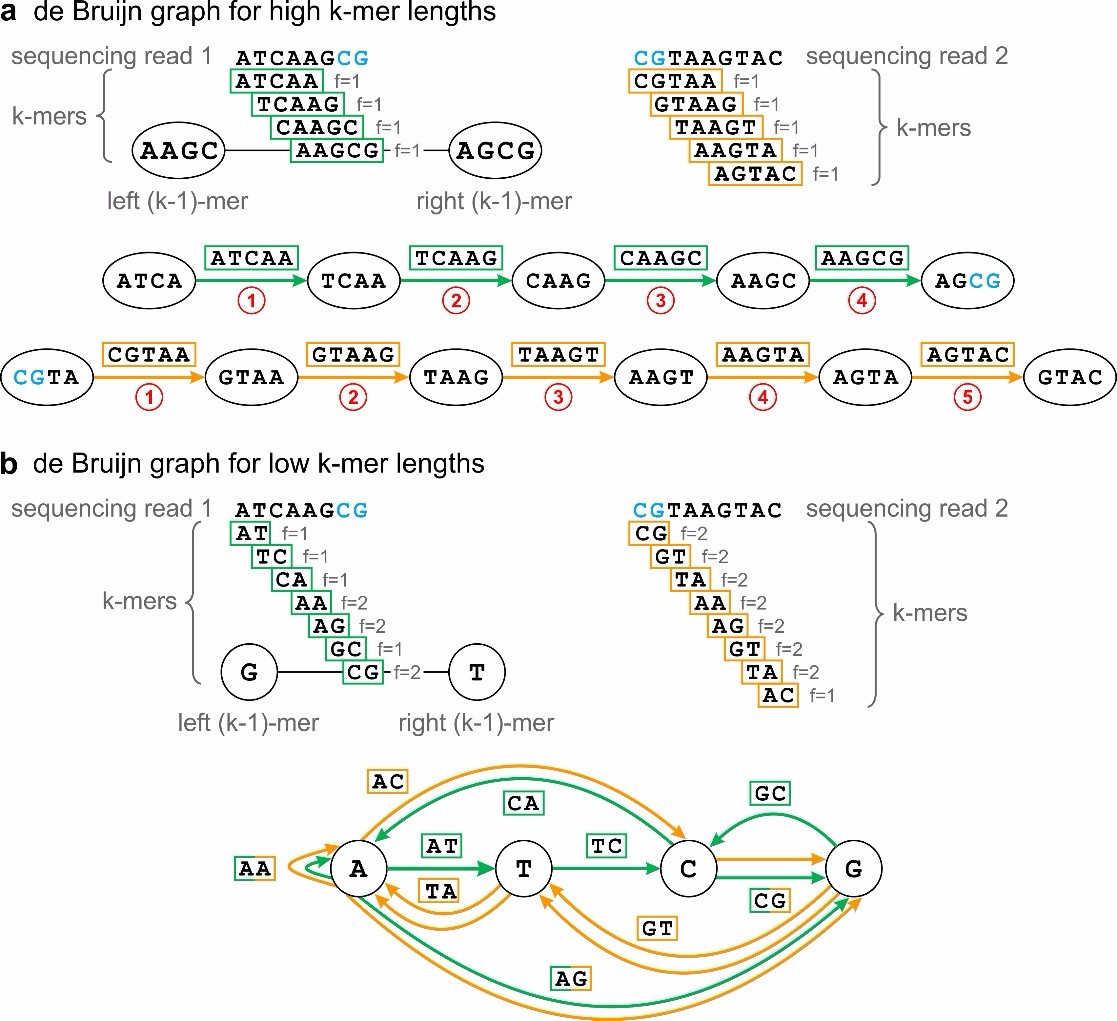
k-mer长度对组装影响
k-mer 长度是不是越短结果越好?
当然不是。因为越短的k-mer,相对应所需要的k-mer之间的碱基配对要求也越低。如果当k=2的时候,那么其实k-mer之间只需要一个碱基配对就能被连接,换句话说,所有的k-mer都能被连接在一起。这明显会增加de Bruijn图的分支和复杂度,对于图形的解读是非常不利的,最终利用短k-mer进行拼接的结果,很有可能是较多的短片段。同时,加之宏基因组中物种之间由于水平基因转移等因素,本来就存在大量的物种间重复序列,如果k-mer过短,很可能造成组装的结果过于片段化,物种间序列也别模糊拼接到一起。
组装时 k-mer 长度多少最好?
其实没有一个固定的标准。因为宏基因组的样本类型复杂,物种丰度与基因组大小飘忽不定,很能设定一个标准。但可以说的是,过短的k-mer不利于物种序列的区分,太长的k-mer不利于对拼接错误的矫正。虽然说得这么玄学,但实际上还有一定方法可以提供组装的质量:
1
在测序深度足够的情况下,尽量选取长度较长的k-mer
2
在样本复杂度不高的情况下,尽量选取长度较长k-mer(其实原理与第一点一样)
3
利用一些新型的组装方法,如MEGAHIT这种选取不同长度k-mer进行迭代的方法,能有效同时兼顾准确性与组装长度。
- 本文固定链接: https://maimengkong.com/kyjc/742.html
- 转载请注明: : 萌小白 2021年8月12日 于 卖萌控的博客 发表
- 百度已收录
