我们可以采用多组学方法分析患病人群肠道菌群的组成与功能差异,进一步阐释微生物与宿主疾病间可能的因果关系,如微生物组学和代谢组学联用。
微生物相关代谢组学可以应用于胃肠病学、生物化学、内分泌学、微生物学、遗传学、营养学、食品科学和药理学等多个领域。过去的十年中,微生物组-代谢组学关联研究数目指数级增长。然而,分析由此产生的多组学数据及其相关性仍然是一个重大的挑战。
目前微生物组-代谢组联合分析策略主要依赖多组学数据间的统计相关性,如Spearman相关性和偏最小二乘判别分析。而Pedersen等人于2018年提出通过WGCNA降维方法来分析宿主表型、肠道宏基因组和代谢组之间的相关性。因此,我们需要一个精简的计算流程来综合分析微生物组与代谢组学数据。
在这里,小编为大家推荐一款可以结合多种单变量、多变量统计方法和相关性分析的微生物组-代谢组联合分析工具:M 2IA。文章于2020年3月份发表在Bioinformatics上,M 2IA:a Web Server for Microbiome and Metabolome Integrative Analysis
(https://doi.org/10.1093/bioinformatics/btaa188 )。
M 2IA拥有基于web的服务器,用户需要上传16S rRNA基因或宏基因组测序技术的微生物组数据,以及质谱或核磁共振光谱平台的代谢组数据来进行下游分析。界面友好且操作简单,即使没有受过生物信息学培训的用户也可以轻松使用。那么大家就跟着小编来一起体验一下吧!
一
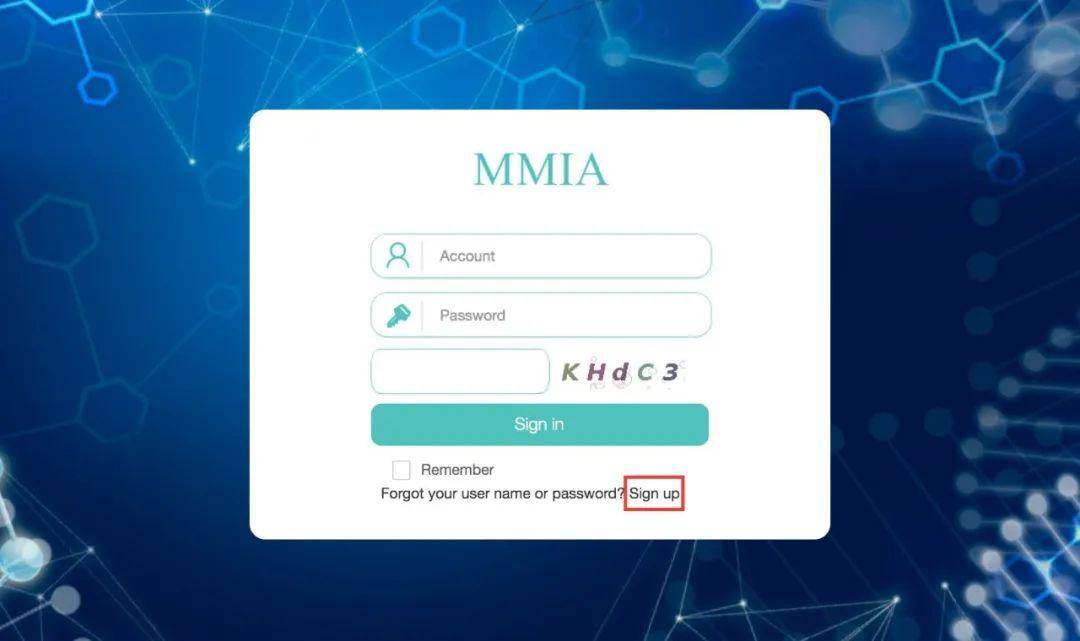
账户注册
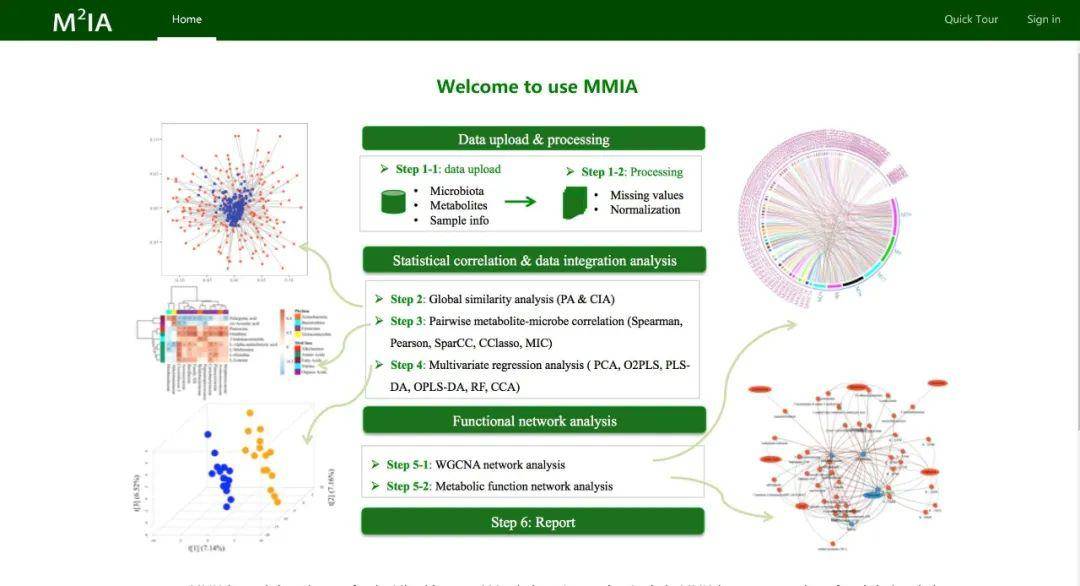
进入网址,我们可以大致了解到它的主要分析功能,如相似性分析、代谢-微生物组相关性分析、多元回归分析(PCA、OPLS-DA、CCA等)以及代谢组的WGCNA、代谢功能网络分析,可以看到示意图还是十分精美的。
首先我们要注册一个账号,这样就可以在网站保存自己的项目和数据。
二
进行项目
我们想要完成一个项目,只需要简单的几个步骤:
1
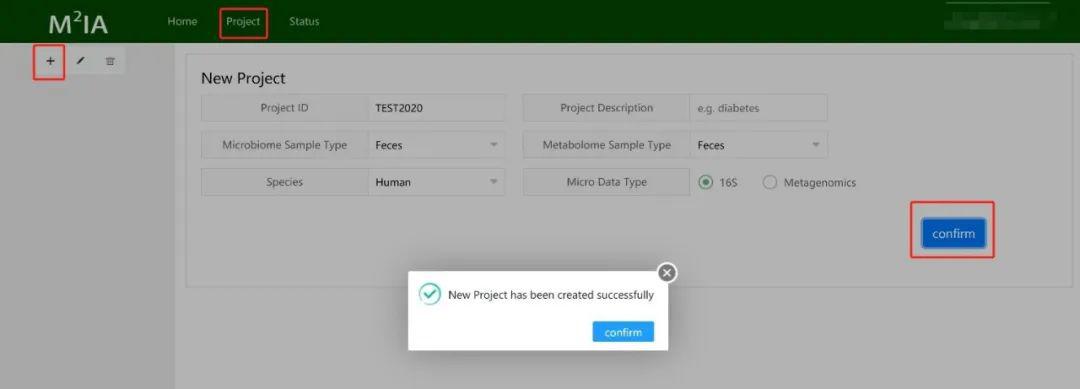
创建项目
首先点击主页面顶部的 ‘project’进入分析页面,然后点击左上角‘+’创建项目,进行确认,即新项目创建完成。
2
上传数据
第二步就是上传我们的数据,需要提交的是微生物组的OUT表、代谢数据以及分组信息表。
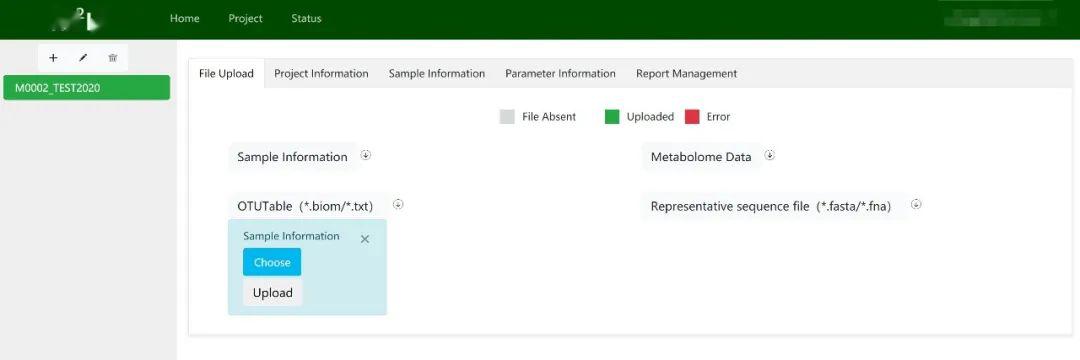
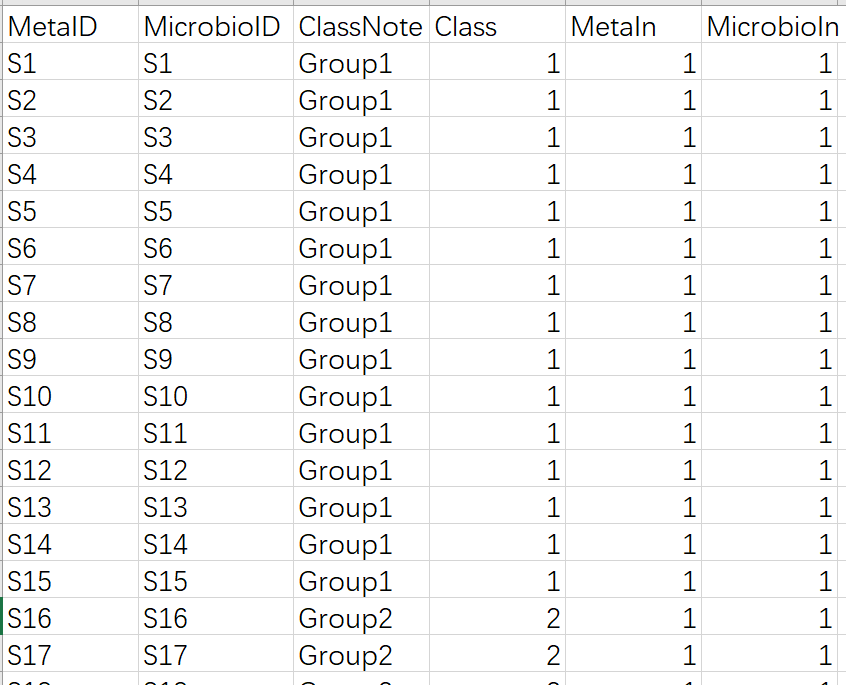
❶对于Sample Information表,就是我们熟知的Metadata文件(原始数据文件),里面包含样本ID以及分组等信息,需要提交CSV格式文件格,分为六列,MetalID即代谢组分析的样本ID,MicrobioID为微生物组的样本ID,ClassNote为样本的分组信息,Class为样本组名,最后MetaIN代表包含代谢组学分析的样本是否需要进行后续分析,是的话为‘1’,否为‘0’。
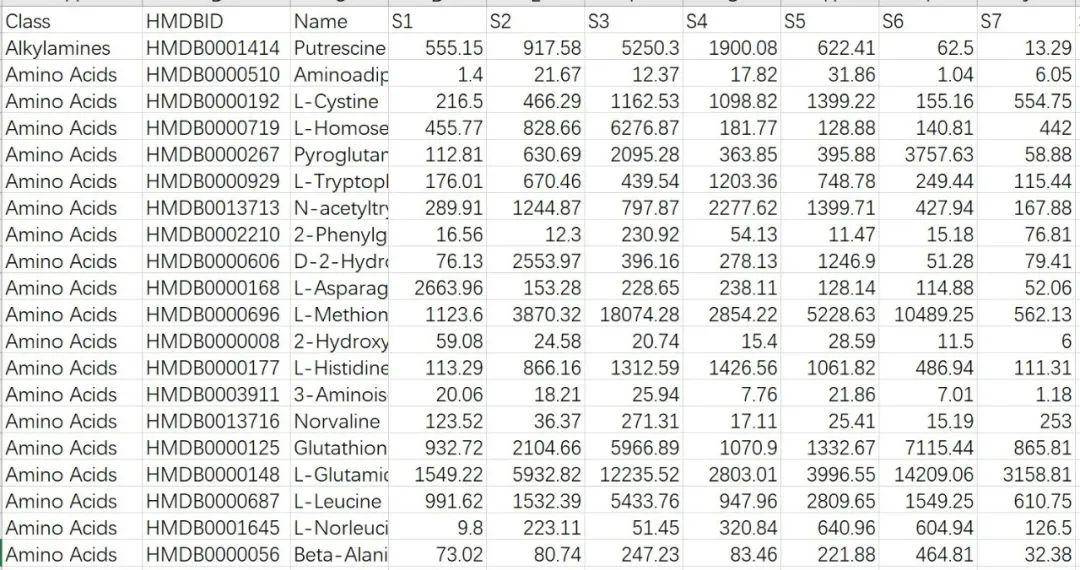
❷代谢组数据
代谢组数据需要提供的是一个CSV格式的文件,前三个中HMDB IDs (Human Metabolome Database,人类代谢组数据库ID)以及代谢物组合名称是必须的。而首列Class为代谢物的分类信息,可以由使用者提供,也可以有M2IA自动匹配生成。The metabolite class information can be provided by users or automatically matched by M 2IA if not provided。其余的列名与之前的样本信息表保持一致即可。
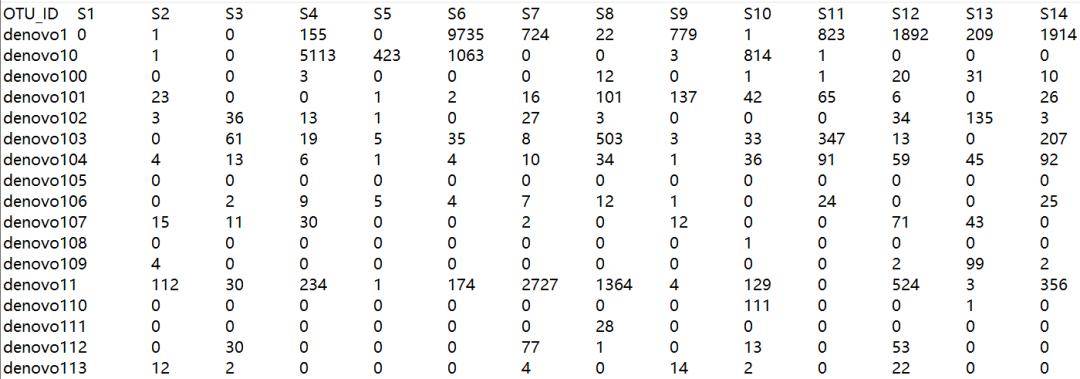
❸ 微生物组数据
使用者提供TXT或者BIOM格式的文件均可,文件需要包含OTU_ID和物种注释信息(taxonomy)。同样,样本ID应该与样本信息表中提供的微生物ID保持一致。
❹代表性序列
这是由微生物组聚类后生成的fasta序列文件,代表性序列ID应该与OTU_ID在OTU表中的保持一致。

3
查看和修改样本信息表
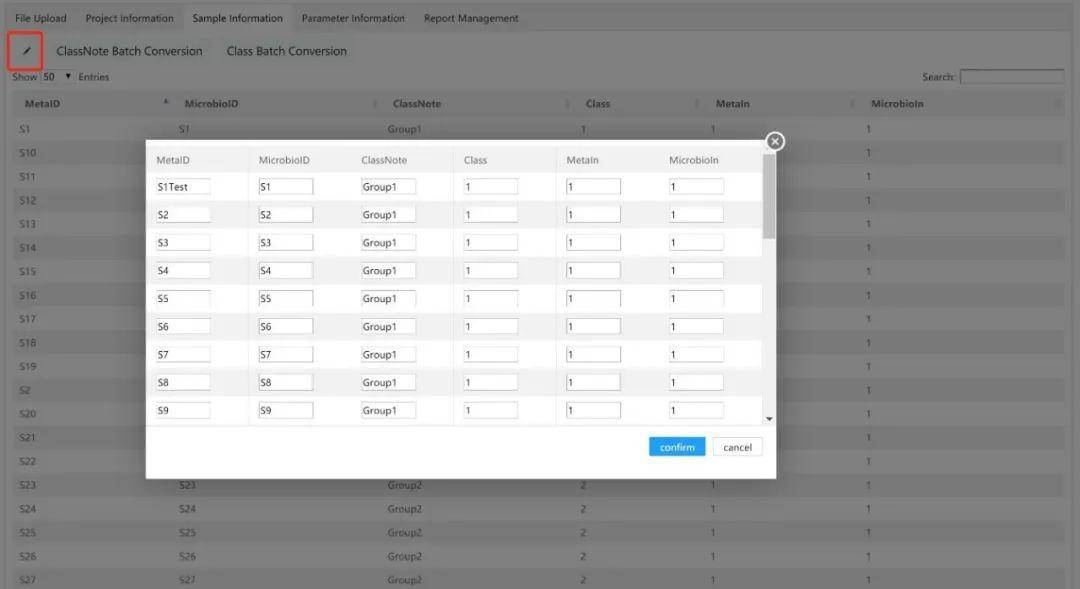
❶在“样本信息”页面,用户可以查看样本信息表。通过单击ClassNote Batch Conversion,用户可以批量修改分类信息。
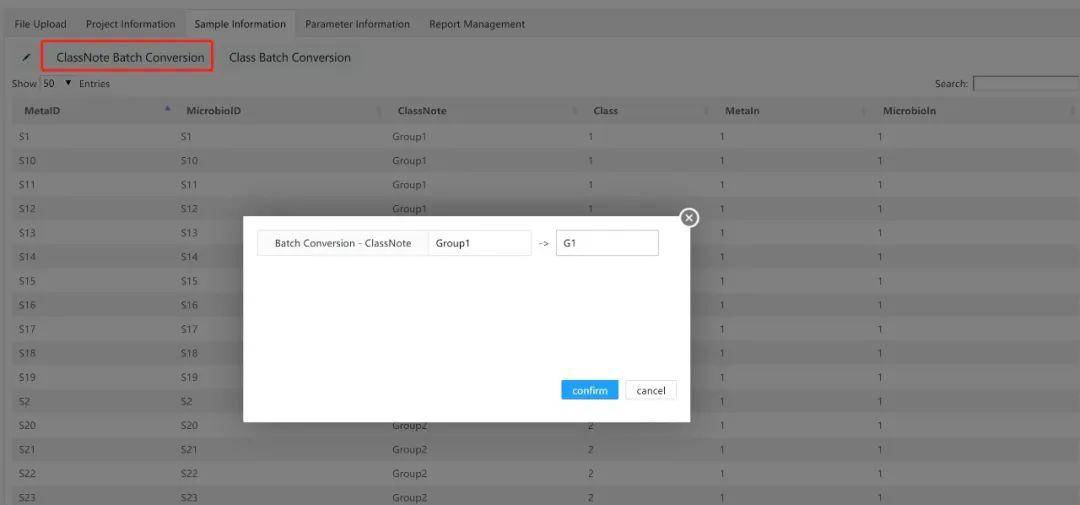
❷ 通过单击修改(modify)按钮,用户可以修改样品信息表任何信息。
4
设置参数
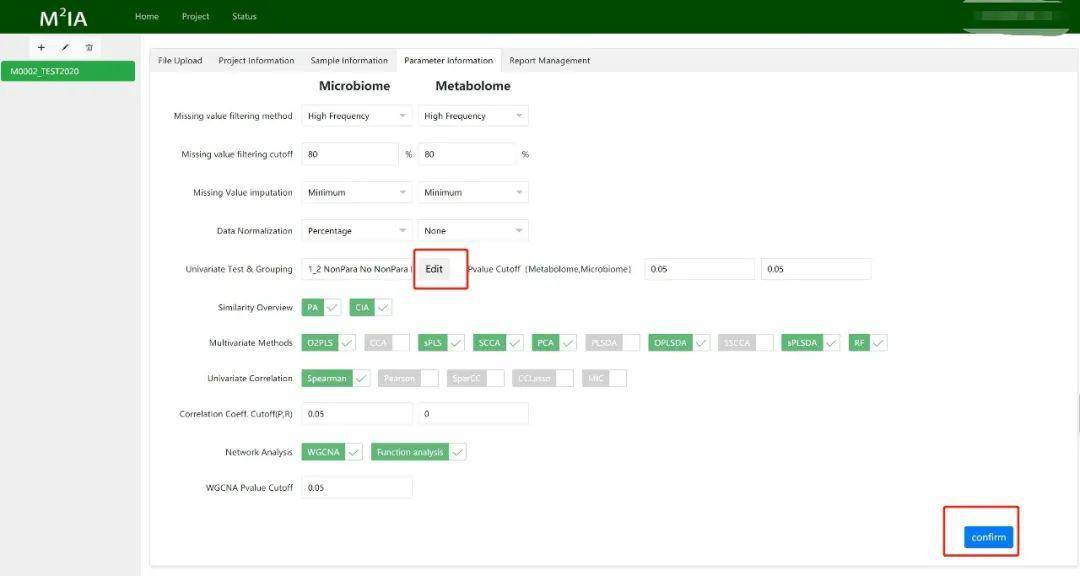
在开始数据分析前我们需要对一些参数进行调整和确认,包括缺失值过滤、标准归一化、单变量检验、相似性分析、多变量分析、相关性分析和功能分析。
用户单击编辑(edit)按钮可以进行单变量检测的设置。在弹出窗口中,用户可以定义需要进行比较的两组,并选择适当的分析方法,例如有参学生t检验或非参Mann-Whitney U检验。选择自动(Auto)选项,程序将通过评估超过60%的变量是否遵循正态分布来自动选择合适检测方法。如果选择了成对(paired)选项, M 2IA将进行成对单变量分析。但是这要求样本ID在两个不同的组(例如A_1和B_1,A_2和B_2)中必须具有相同的后缀。
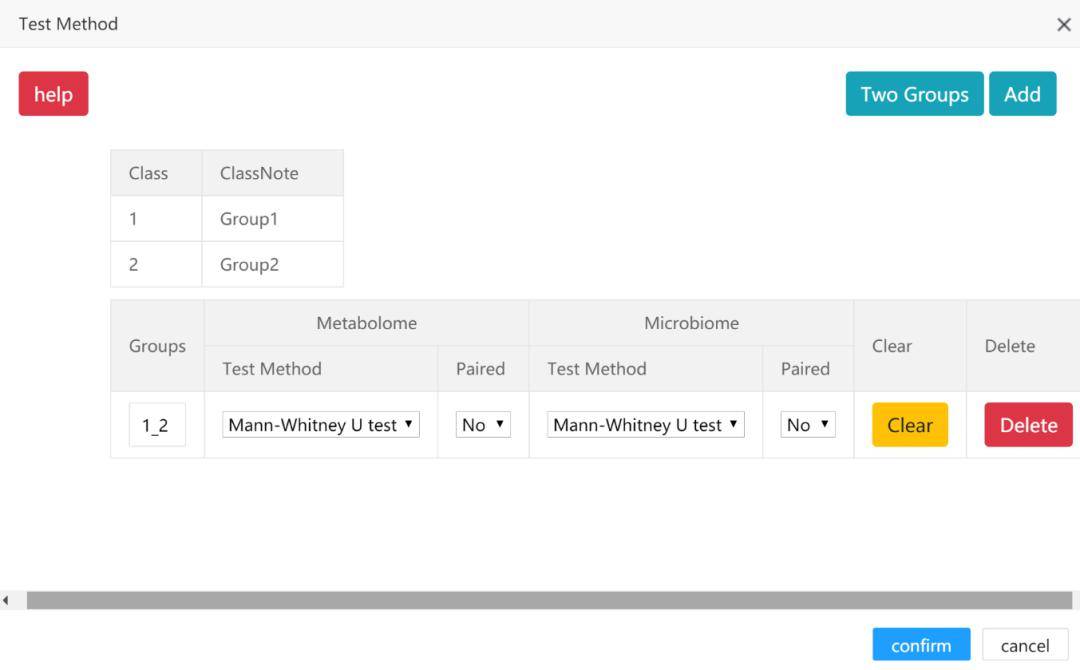
最后,用户需要点击确认即可。
5
项目提交
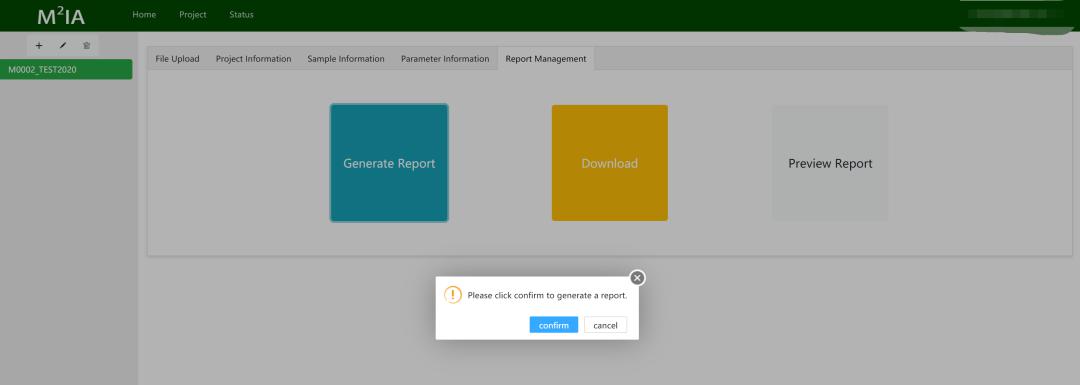
进入项目管理页面,点击生成报表(Generate Report )按钮提交作业,开始数据分析。同时,“预览报告”按钮是灰色的,直到报告生成。
6
检查进度
进入状态页,用户可以查看项目提交的状态。
7
检查结果报告
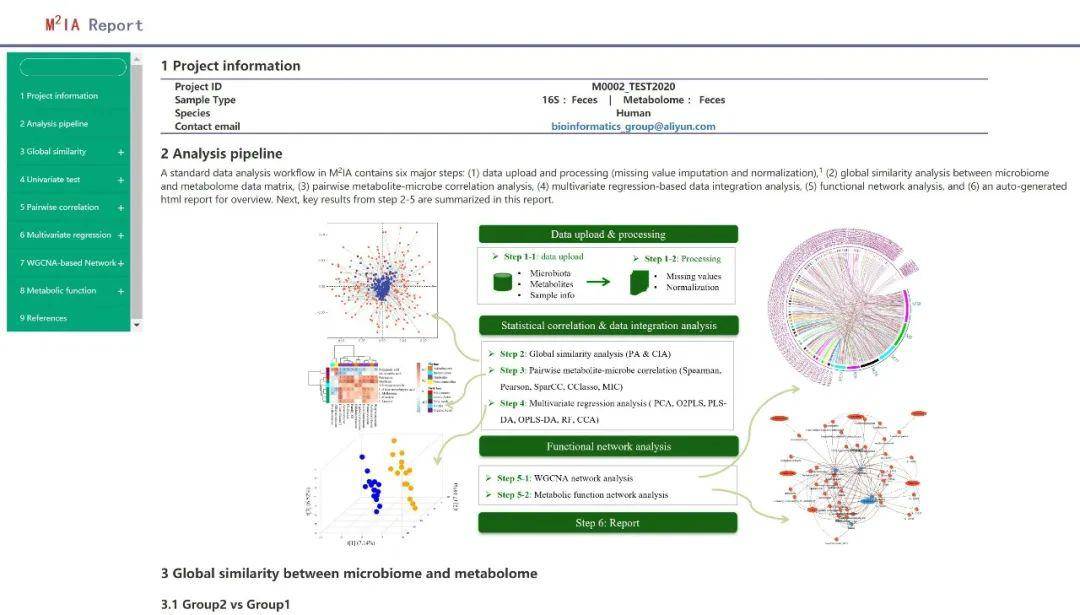
当预览报告(Preview Report) 按钮变为绿色时,数据分析就完成了,并成功创建了报告摘要。点击Preview Report ,用户即可以在线预览分析结果。(注意:不活跃账户和数据只可以在网站保留三个月)
8
下载结果Download Results
压缩格式的数据表和图可以直接到下载(download)页面进行下载。小编就为大家展示一下示例数据的分析结果吧。
示例数据使用的是TwinsUK 团队2018年发表在Gut Microbes是关于人体内脏脂肪相关肠道菌群的部分数据,研究发现肠道菌群苯甲酯降解和磷酸转移酶系统的代谢途径与肥胖密切相关。
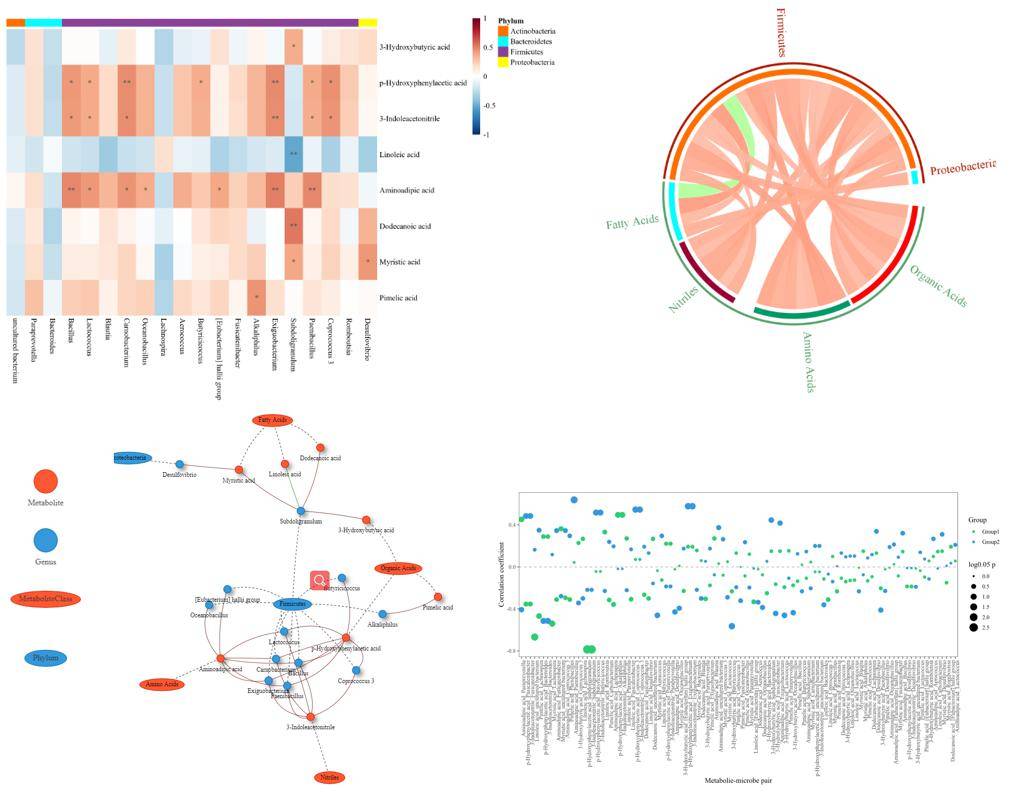
图1:热图、圈图、网络图、点图展示代谢组、微生物组相关性
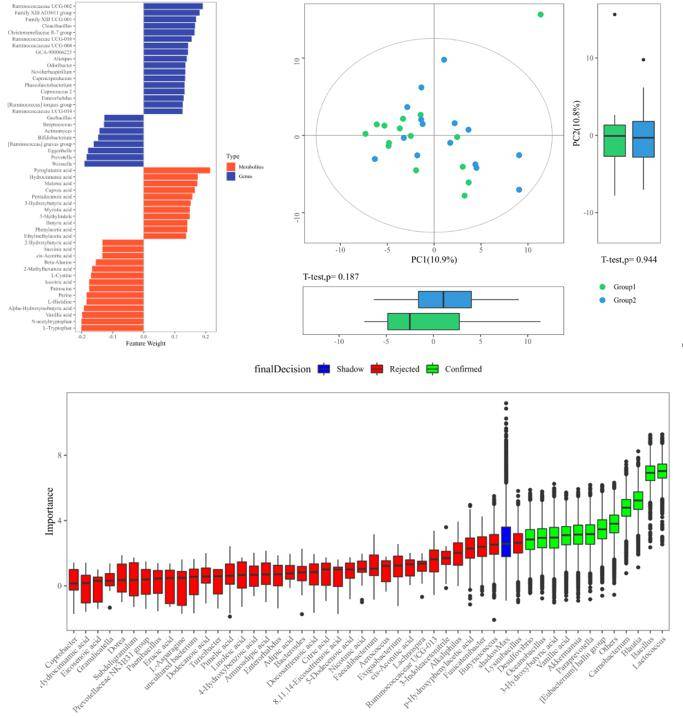
图2:多元回归分析:
三
总 结
M 2IA是一款方便易用的微生物组联合代谢组数据下游网页分析展示工具,注册后既可免费使用。利用WGCNA来检测宿主表型、肠道宏基因组和代谢组之间的相关性,提供用户友好的图形界面,集成了多种先进的统计方法,使整个数据分析过程自动化,并可以一键生成详细的数据解释报告。在分析平台方面,M 2IA接受来自16S rRNA基因测序或宏基因组测序技术的微生物组数据,以及来自质谱或核磁共振分析平台的代谢组数据。建议使用同一主体的人类粪便样本或动物粪便/结肠内容物来检测微生物组和代谢组。
缺点:M 2IA接受带注释的微生物和代谢物作为输入数据,但无法对原始数据进行预处理,如微生物组数据的质控和物种注释,代谢组数据的峰值检测和代谢物注释。目前已有多种用于原始数据预处理的软件工具或管道,如用于16S rRNA测序的QIIME2和基于MS代谢的XCMS。M 2IA的重点是提供一个标准化和全面的代谢组学联合微生物组学数据的综合分析流程,探讨微生物及代谢物在统计学意义上的相关性以及相互作用网络。此外,基于KEGG数据库的功能网络分析有助于解释它们的生物学相关性,并建立适用于开发个性化治疗的机制假设(如益生菌、生物制剂的开发)。
这款工具可以说是微生物组科研工作者的福音,可以替代大部分需要代码完成的下游分析工作,同时一键生成分析报告,用于同行交流或者撰写文章。当数据需要再深度挖掘时,此工具也可以作为前期数据的准备,强烈推荐,大家可以自行探索使用。
- 本文固定链接: https://maimengkong.com/kyjc/1711.html
- 转载请注明: : 萌小白 2024年4月1日 于 卖萌控的博客 发表
- 百度已收录
