1. 概述
1.1. 背景及分析流程简介
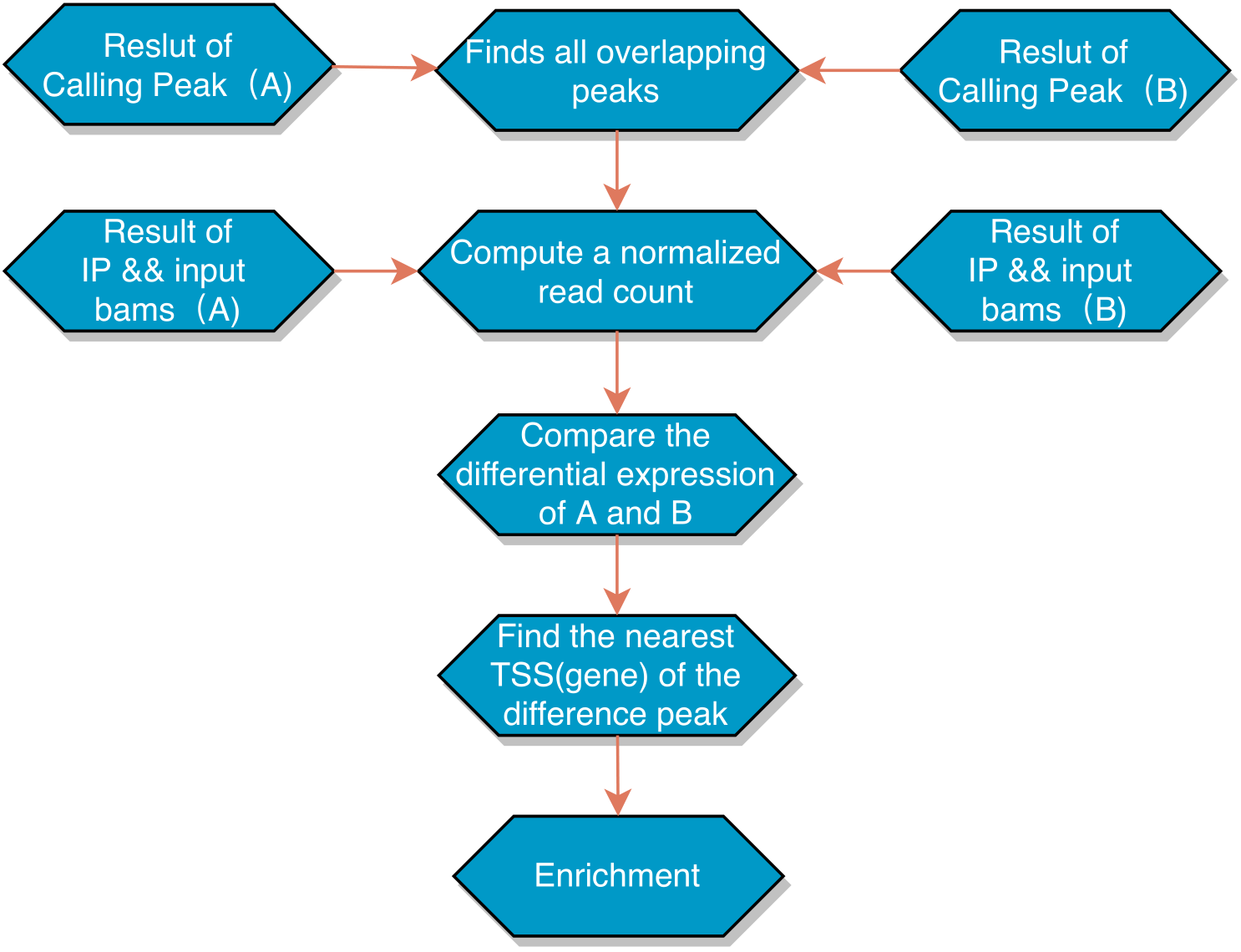
为了理解细胞中更为复杂的生物过程,许多研究已在通过比较ChIP-seq的差异获得的不同数据。越来越多的ChIP-seq实验正在研究多种实验条件(例如各种治疗条件,几个不同的时间点和不同的治疗剂量水平)下的转录因子结合,组蛋白修饰的差异。差异富集在生物学和医学研究中已变得具有实际重要性。 为了建立对比条件消除误差,我们对数据进行了以下流程处理:我们首先将A与B两组的结果进行共有Peak区域基因计算,对于有共有区域(overlap)的Peak,计算最高峰位点并向其两侧各延伸250bp作为合并峰计算区域,对每个区域进行的每组样本进行reads表达定量,进行差异Peak的计算,筛选出差异Peak,进行临近3K注释到基因上,进行基因集富集分析。
本组实验结果,我们处理的是有两组重复的DiffPeak数据对比,我们的差异Peak筛选标准为:|log2FC| > 1 && FDR < 0.05。
分析流程:
1.2. 结果汇总
| 路径 | 说明 |
|---|---|
差异Peak分析结果, 目录: Results/
|
|
Results/*DiffPeakInfo.xls
|
差异Peak计算的所有结果 |
Results/*DiffPeakInfo.bed
|
差异Peak计算的所有结果的bed文件 |
Results/*DiffPeakInfo_FC2-q0.05.xls
|
差异Peak计算结果按阈值筛选后结果 |
Results/*DiffPeakInfo_FC2-q0.05.bed
|
差异Peak计算结果按阈值筛选后结果的bed文件 |
Results/*DiffPeakInfo_FC2-q0.05_GAIN.bed
|
差异Peak计算结果按阈值筛选后结果的bed文件(差异上调) |
Results/*DiffPeakInfo_FC2-q0.05_LOSS.bed
|
差异Peak计算结果按阈值筛选后结果的bed文件(差异下调) |
Results/*DiffPeakInfo_FC2-q0.05_PeakAnno.xls
|
差异Peak计算结果按阈值筛选后结果的临近注释文件 |
Results/*DiffPeakInfo_FC2-q0.05_PeakAnno.sorted.xls
|
同上,差异Peak计算结果按阈值筛选后结果的临近注释文件 (按annotation(Promoter), Fold, FDR列排序) |
Results/*DiffPeakInfo_FC2-q0.05_PeakAnno_gene.bed
|
注释到的基因(转录本)信息标记bed文件 |
差异Peak分析绘图结果, 目录: Results/Plot
|
|
Results/Plot/1cor_peakScore_*.png
|
peak相关性热图分析 |
Results/Plot/1pca_peakScore_*.png
|
peak相关性PCA分析 |
Results/Plot/2cor_readCount_*.png
|
共有区域的readCount相关性热图分析 |
Results/Plot/2pca_readCount_*.png
|
共有区域的readCount相关性PCA分析 |
Results/Plot/*_1cor.png
|
差异Peak相关性热图分析 |
Results/Plot/*_2pca.png
|
差异Peak的PCA分析 |
Results/Plot/*_3ma.png
|
差异Peak的MA图 |
Results/Plot/*_4vol.png
|
差异Peak的火山图 |
Results/Plot/*_5box.png
|
差异Peak的箱型图 |
Results/Plot/*_6heatmap.png
|
差异Peak的热图 |
显著差异Peak的临近基因集富集分析, 目录: Results/Enrich/
|
|
Results/3.Enrich/*/
|
各组差异Peak的临近注释基因集的富集分析结果目录 |
Results/3.Enrich/*.html
|
辅助查看富集结果的网页文件 |
Results/3.Enrich/*/*-p.adjust1.00.csv
|
富集分析结果列表(原始) |
Results/3.Enrich/*/*-p.adjust0.05.csv
|
富集分析结果列表(按padj<0.05筛选后) |
Results/3.Enrich/*/*.pdf
|
富集分析的绘图高清文件 |
* 以上重要结果为加粗显示。
2. 分析流程
2.1. 重叠区域的计算
2.1.1. PeakScore相关性分析
为了进行后续的差异Peak的富集程度比较,我们需要合并Peak比较区域,在overlap的共有区域计算前,我们需要先了解各组内的peak重复性情况。 对Treat组和Control组进行PeakScore相关性热图分析,PCA分析。
|
Results/Plot/1cor_peakScore_Demo_A-B.png 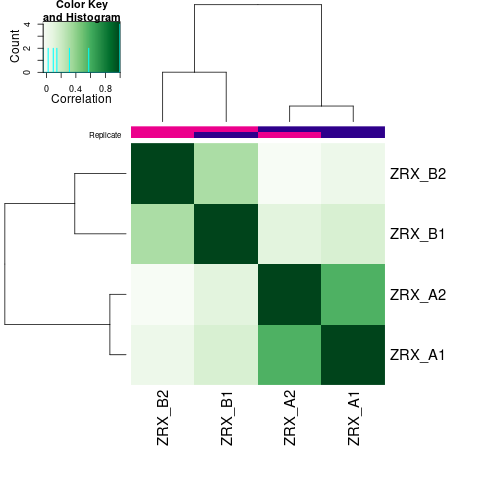
|
Results/Plot/1cor_peakScore_Demo_C-D.png 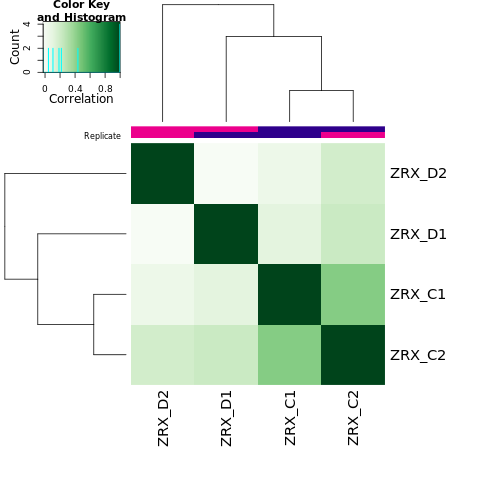
|
|
Results/Plot/1pca_peakScore_Demo_A-B.png 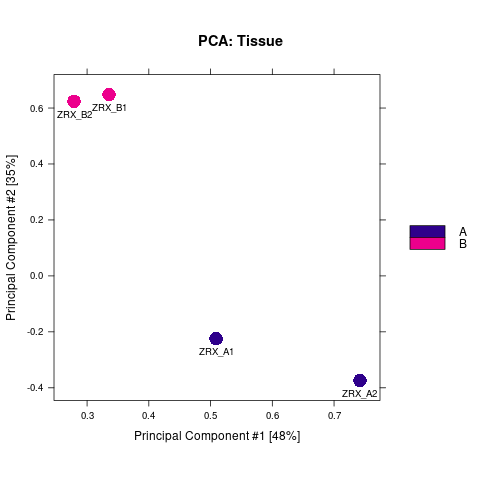
|
Results/Plot/1pca_peakScore_Demo_C-D.png 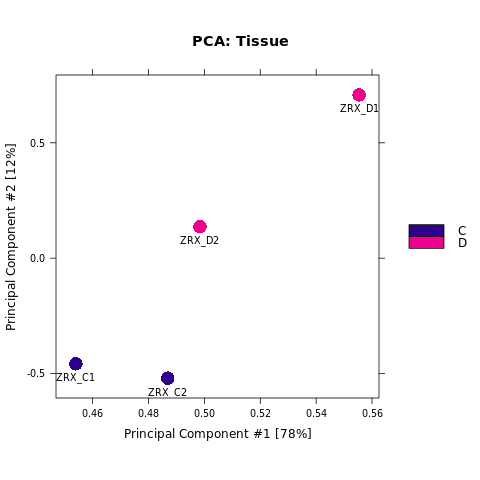
|
2.1.2. readsCount相关性分析
我们选取至少含有overlap区域>=2个样本的callPeak区域结果,计算最高峰位点并向其两侧各延伸250bp作为合并峰计算区域,对每个区域每组样本进行reads表达定量。 随后,我们对各组进行readsCount的相关性热图分析,PCA分析。
|
Results/Plot/2cor_readCount_Demo_A-B.png 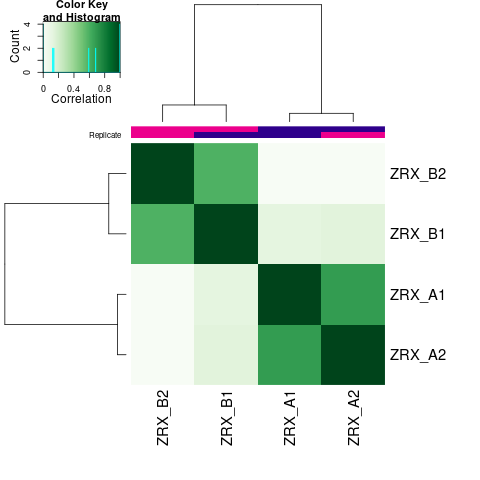
|
Results/Plot/2cor_readCount_Demo_C-D.png 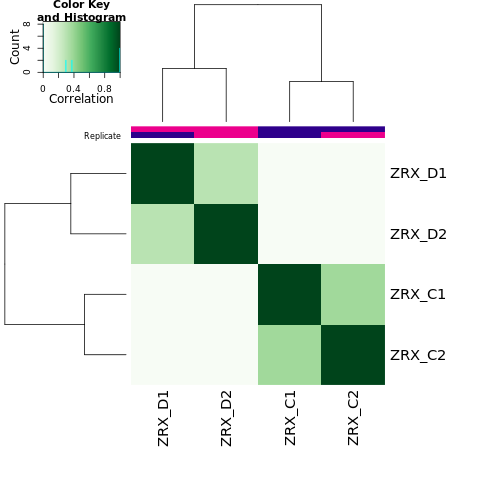
|
|
Results/Plot/2pca_readCount_Demo_A-B.png 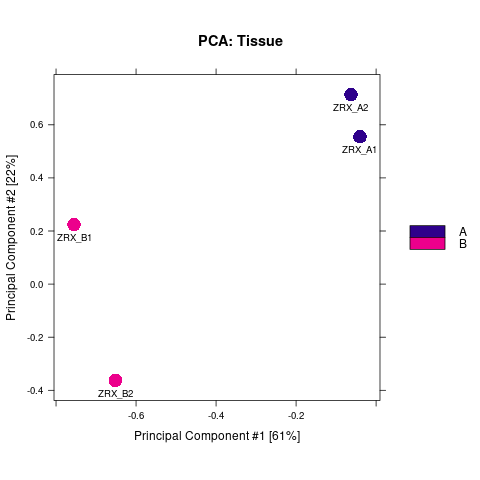
|
Results/Plot/2pca_readCount_Demo_C-D.png 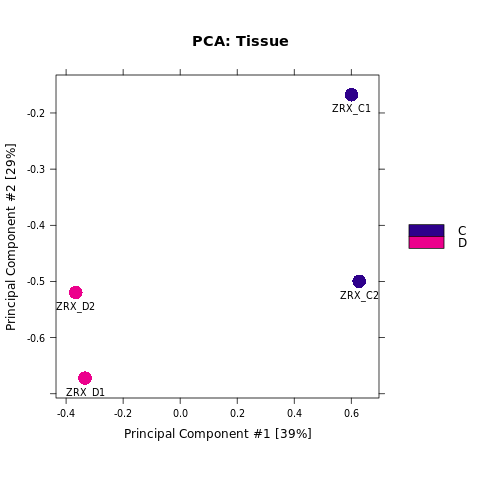
|
2.2. 差异Peak的计算
2.2.1. 差异Peak的相关性计算及显著性差异Peak的筛选
通过计算两组之间的合并区域的表达差异,我们能获得两组比较计算的差异Peak所有结果。 通过相关性热图及PCA,可以看出组内的差异peak计算的相关性好坏,一般而言好的结果能明显区分开。 通过阈值|log2FC| > 1 & FDR < 0.05进行筛选获得显著差异Peak筛选结果,进行相关性热图,PCA,火山图,热图绘制如下。
通过差异Peak分析,我们得到了基因组范围内的差异Peak信息,为进一步得到差异Peak附近的临近基因信息,我们使用Chipseeker进行进一步注释,得到Peak所对应的临近注释基因,并给出Peak在Promoter的上下游3k,或之外的Intron、Exon等区域的位置及距离等信息的注释文件: Results/*DiffPeakInfo_FC2-q0.05_PeakAnno.xls。
|
Results/Plot/Demo_A-vs-B_1cor.png 
|
Results/Plot/Demo_C-vs-D_1cor.png 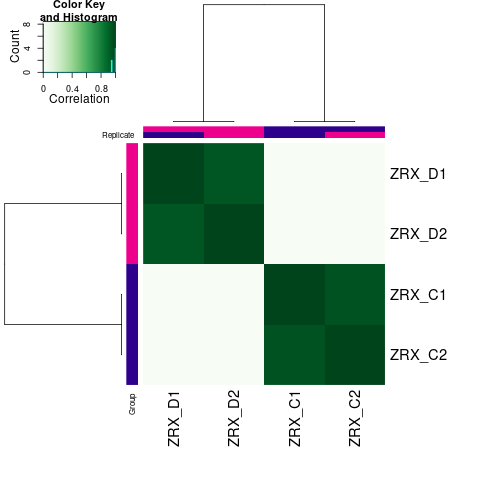
|
|
Results/Plot/Demo_A-vs-B_2pca.png 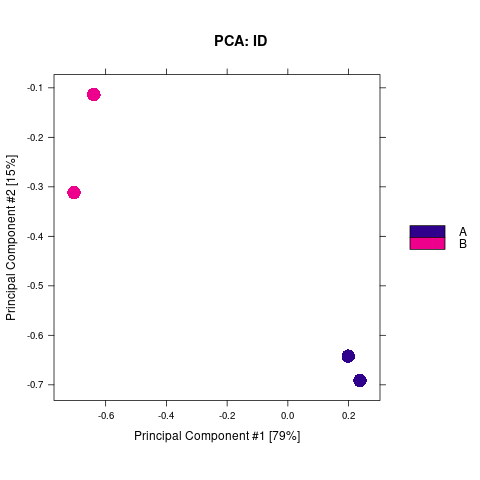
|
Results/Plot/Demo_C-vs-D_2pca.png 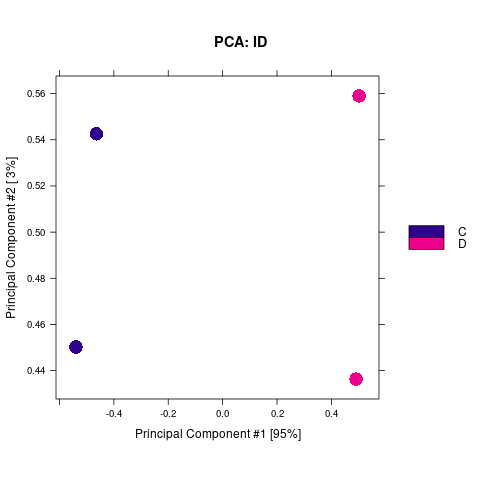
|
|
Results/Plot/Demo_A-vs-B_4vol.png 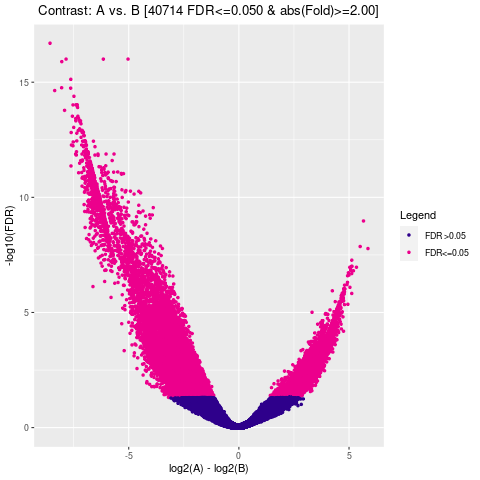
|
Results/Plot/Demo_C-vs-D_4vol.png 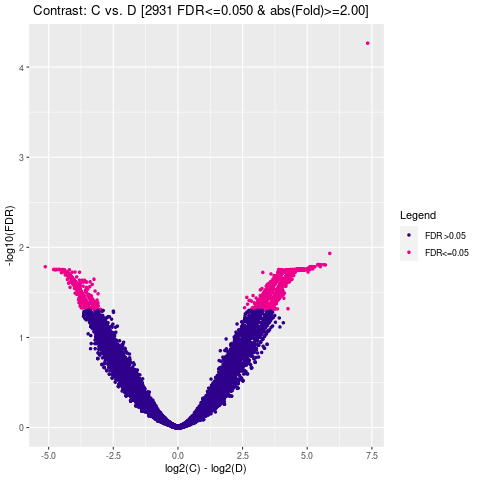
|
|
Results/Plot/Demo_A-vs-B_6heatmap.png 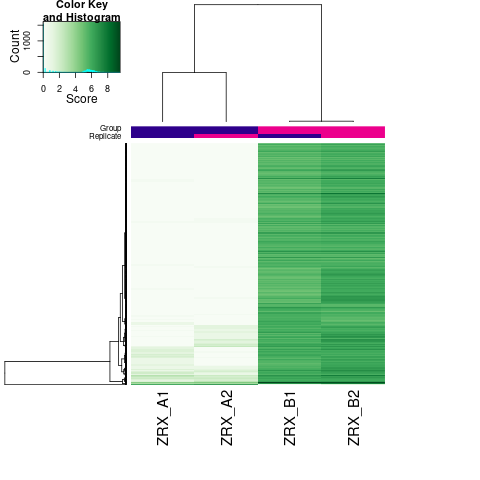
|
Results/Plot/Demo_C-vs-D_6heatmap.png 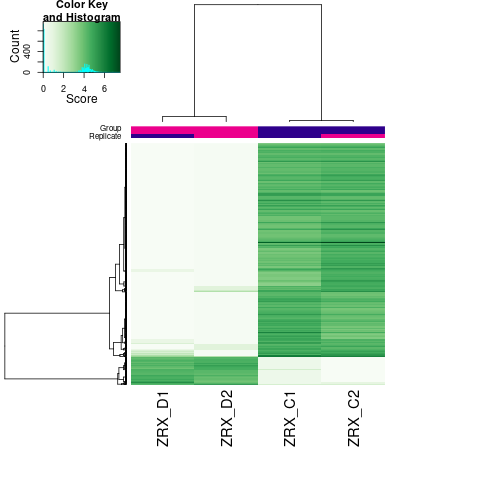
|
Results/*DiffPeakInfo_FC2-q0.05_PeakAnno.xls表头说明:
| 表头 | 说明 |
|---|---|
peakname
|
差异Peak的name |
seqnames
|
差异Peak所在染色体 |
start
|
差异Peak在参考序列上的起始位置 |
end
|
差异Peak在参考序列上的终止位置 |
width
|
差异Peak的长度信息 |
strand
|
正负链信息 |
Conc
|
Group1和Group2平均值进行log2标准化后的计数 |
Conc_Group1
|
Group1进行log2标准化后的计数 |
Conc_Group2
|
Group2进行log2标准化后的计数 |
Fold
|
Group1与Group2的差异倍数(进行log2标准化) |
p.value
|
差异Peak的置信度计算 |
FDR
|
差异Peak的多重校验FDR |
change
|
上下调标记,上调标记为GAIN,下调标记为LOSS |
annotation
|
peak注释信息(对于注释到基因上等注释信息的描述) |
geneChr
|
注释基因的染色体信息 |
geneStart
|
注释基因的起始位置 |
geneEnd
|
注释基因的终止位置 |
geneLength
|
注释基因的长度 |
geneStrand
|
注释基因的正负链 |
geneId
|
注释基因的EntrezID |
transcriptId
|
注释基因的转录本名字 |
distanceToTSS
|
被注释Peak距离TSS的距离 |
ENSEMBL
|
注释基因的ENSEMBL名 |
SYMBOL
|
注释基因的SYMBOL名 |
GENENAME
|
注释基因的基本描述信息 |
2.2.2. 差异Peak注释基因的富集分析
将上述临近注释得到的基因集,进一步进行GO和KEGG富集分析,得到差异Peak筛选结果的临近注释基因富集结果。结果文件说明及解读,同CHIP标准分析流程报告。
结果目录: Results/Enrich/
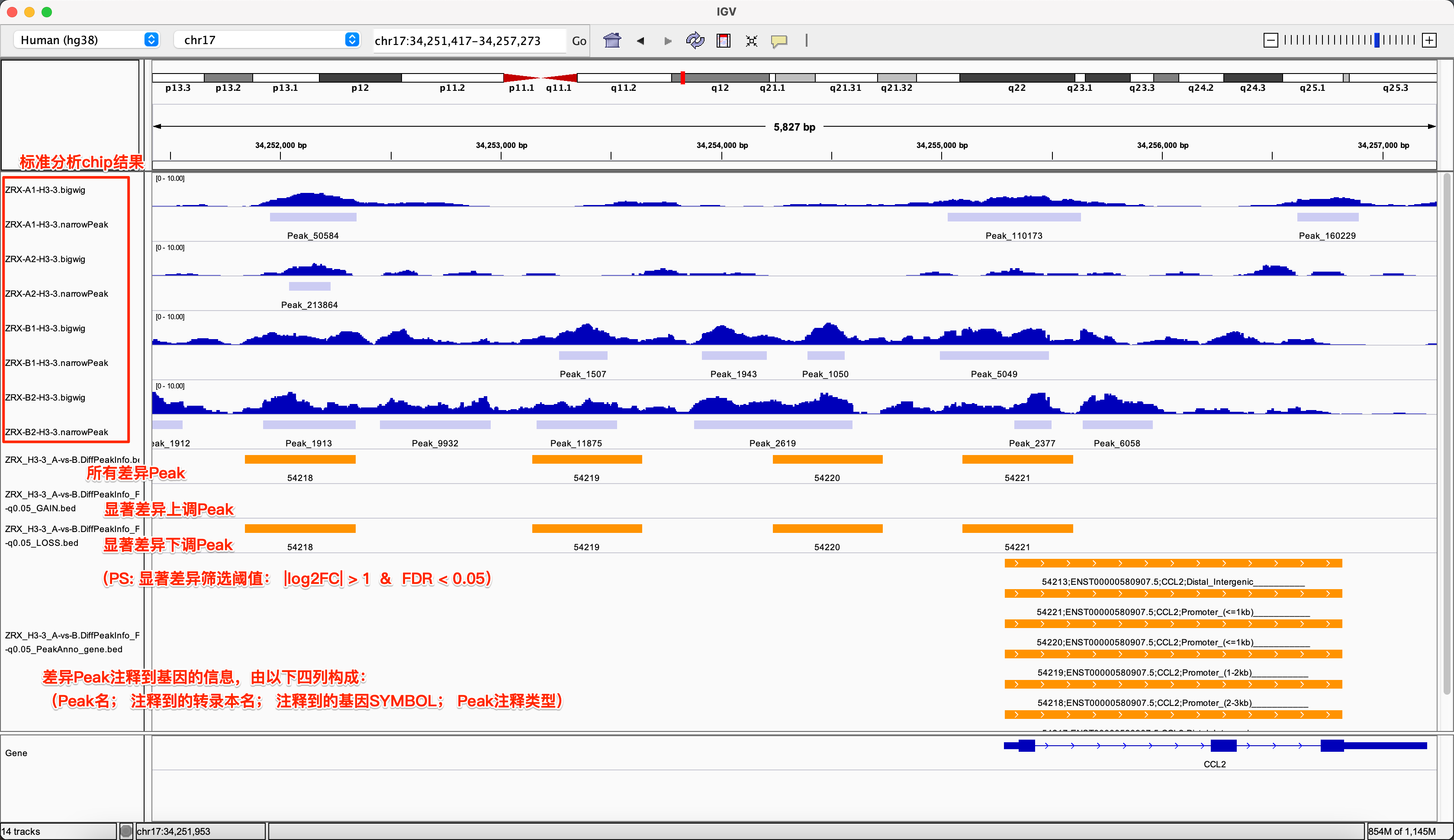
3. 结果的IGV可视化
为了得到较为直观的测序分析结果,我们一般需要借助可视化工具,IGV在这个过程中扮演十分出色的角色,他不仅展示了不同样本测序覆盖情况,还常常用于联合分析,如mRNA的测序变化与chip测序的变化。 在此项目中,我们用于差异Peak的筛选与评估,我们可将分析结果文件导入,步骤如下:
-
导入CHIP分析结果,即前面我们的Chip标准分析结果中
.bigwig与.narrowPeak文件。 -
导入CHIP的差异Peak分析结果,即本分析中所得到的bed结果。
-
调节数据显示范围:
-
bigwig 高度范围显示调节:按住
ctrl / command选中多个.bigwig文件,右击点击Set Data Range...。 为方便对比,在对比不同区域Peak时,可手动调节显示范围。 -
bed / gene 重叠区域展开设置: 右击bed文件,点击
Expanded设置展开。 -
搜索感兴趣的
Peakname / SYMBOL: 在第一排第三个框内输入Peakname / SYMBOL名,点击GO即可搜索。如果搜索不到,可尝试点击Reload重新加载。
筛选的 Peakname / SYMBOL 的一些方法:
-
搜索感兴趣的Peak,可参考:
Results/*DiffPeakInfo_FC2-q0.05_PeakAnno.sorted.xls,该文件按annotation(Promoter), Fold, FDR列排序, 即Promoter上游3K区域差异倍数较大的结果将被优先排序。 排名较前的结果具有一定的显著差异Peak筛选价值。 -
搜索感兴趣的Gene,可根据生物学功能研究,挑选出较有意义的功能富集结果的基因集,反向去看差异Peak变化情况。 上述分析的功能富集结果具有一定的参考意义。
Demo展示:
一个示例如下,在该IGV中通过可视化,可读出的信息有:在 A vs B 的差异Peak对比中, Peakname 为 54218, 54219, 54220, 54221 的这些Peak比较区域, A相对B具有显著下调趋势,它们都被临近注释到CCL2基因上,注释类型为3K内的Promoter。
示例图:
- 本文固定链接: https://maimengkong.com/kyjc/1584.html
- 转载请注明: : 萌小白 2023年6月22日 于 卖萌控的博客 发表
- 百度已收录
