前言:后天我们的多组学文献精读会分享一篇有关 染色质可接近性的文章,考虑到大部分人可能对其中的一些概念还不了解,所以今天我们就来提前学习一下有关ChIP-seq和ATAC-seq的一些基础知识。
(视频讲者:张健教授团队张月明)
主要内容
- 启动子、增强子的概念
- 什么是基序(motif)
- 什么是染色质可接近性
- 什么是组蛋白修饰
- 研究染色质可接近性的测序技术
01
启动子、增强子的概念
增强子和启动子的关系:增强子是通过启动子来增加转录的。
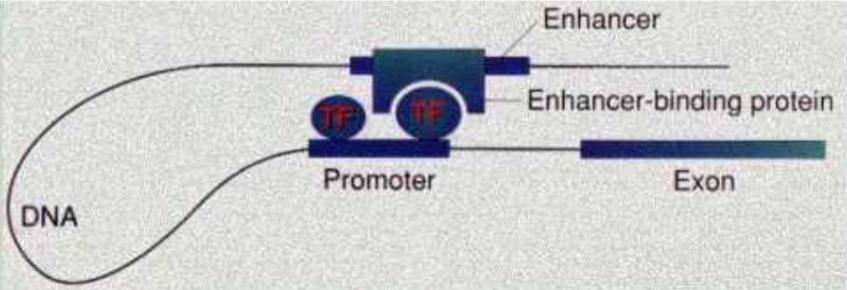
增强子和启动子之间的相互作用
有效的增强子可以位于基因的5’端,也可位于基因的3’端,有的还可位于基因的内含子中。可以看到增强子和调控的基因的关系相对于启动子和其调控的基因要复杂得多。所以在下周的另一篇有关染色质可接近性的文章中,作者预测增强子和基因之间的关联时用到了一项技术——HiChIP。
在这里我们就简单介绍一下什么是HiChIP:以启动子和增强子为例,我们已经知道他们俩是可以通过转录因子这个蛋白产生相互作用的,我们用一些试剂将他们这种相互作用在细胞内固定下来,然后将染色质打断,加上特定的接头,使这些断裂的末端可以相互连接,然后再用靶向目标转录因子的抗体将他们拉下来,将DNA和蛋白分离,送DNA去测序,我们就可以知道哪些染色质之间存在相互作用了。也就是说,知道了增强子和启动子之间的联系,又通过启动子和靶基因的联系,我们就可以知道增强子可以调节哪些基因了。
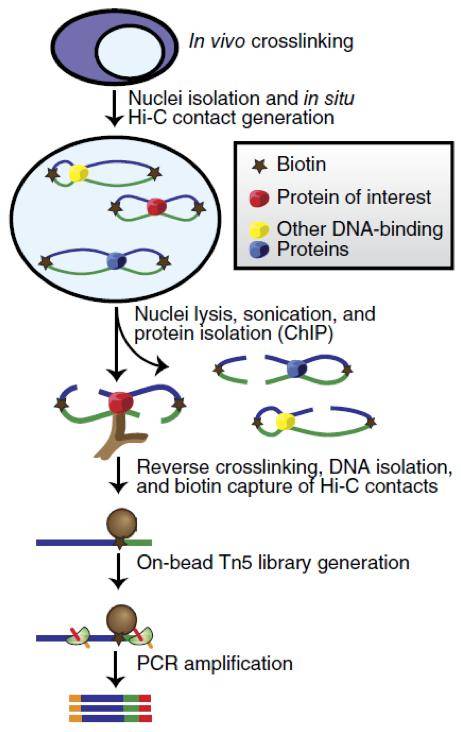
HiChIP的原理图
02
什么是基序(motif)
转录因子在与DNA序列结合时,其结合位点的序列是具有一定的特异性的。个人认为基序可以理解为不同转录因子结合的DNA序列的一个模式。为什么说是一个模式?因为基序并不是特指某一段DNA的具体序列信息。如下图所示是同一个转录因子在多个基因上的结合位点的序列信息:
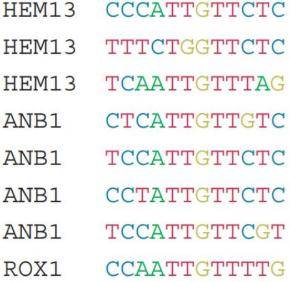
如果我们要用一段序列来描述以上所有的序列的碱基组成,我们就把这一段序列称之为一致性序列。既然如此,我们就需要一个统一的规则来进行这个转换。下图是IUPAC标准的碱基表示法,不同字母对应的碱基如下所示:
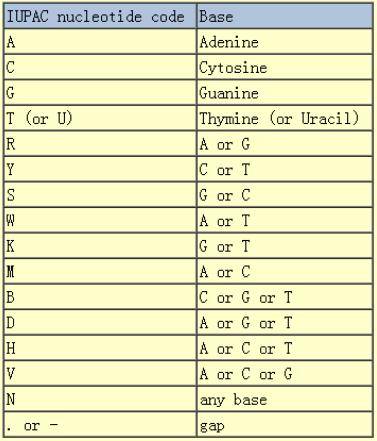
有了这个规则,我们就可以将上述例子表示为如下的一致性序列,也就是我们理解的基序(motif):
03
什么是染色质可接近性
染色体/质的结构:真核生物的核DNA并不是裸露的,上面有组蛋白相结合。DNA一圈一圈地缠绕在组蛋白上,形成串珠式的结构。进一步折叠、浓聚,便形成染色体,不具有转录活性。
染色质可接近性:DNA的复制和转录,都需要将DNA的高级结构打开。但是并不需要将整个DNA全部打开,只需要打开需要表达基因的那一部分即可。而这一个过程,主要由染色体组蛋白的修饰来实现的。这部分松散的染色质被称为开放染色质(open chromatin)或可接近性染色质(accessible chromatin)。染色质的可接近性也可以翻译为可及性或可获得性。染色质一旦打开,就允许一些调控蛋白(比如转录因子和辅因子)与之相结合,并发挥相应的作用。
04
什么是组蛋白修饰
组蛋白包含五个组分,按照分子量由大到小分别称为H1、H3、H2A、H2B和H4。 组蛋白修饰是指组蛋白在相关酶作用下发生甲基化、乙酰化、磷酸化、腺苷酸化、泛素化、ADP核糖基化等修饰的过程。
我们需要简单了解以下4点:
- H3·H4 的乙酰化可形成一个开放的染色质结构, 增加基因的表达;
- 组蛋白甲基化修饰多发生在H3、H4,与基因抑制及基因的激活相关,取决于被修饰的位置和程度;
- 组蛋白磷酸化修饰一般与基因活化相关;
- 组蛋白泛素化修饰则是启动基因表达。
05
研究染色质可接近性的测序技术
在这里,我们主要讲解4种相关的测序技术: ChIP-seq、DNase-seq、MNase-seq、ATAC-seq。当然除了这四种技术,也还有其他的,感兴趣的可以自行了解。
ChIP-seq
ChIP全称:染色质免疫共沉淀(Chromatin immunoprecipitation ,ChIP),seq表示测序。
原理:首先通过染色质免疫共沉淀技术(ChIP)特异性地将目的蛋白及其结合的DNA片段一起拉下来,并对DNA进行纯化与文库构建;然后对富集得到的DNA片段进行高通量测序。再将获得的数百万条序列精确定位到基因组上,从而获得全基因组范围内与组蛋白、转录因子等互作的DNA序列信息。
ChIP的实验流程:DNA与蛋白质交联→超声裂解→免疫沉淀→PCR扩增→测序(如下图所示)
ChIP的分析流程:
(1) 质控
(2) 序列比对:将原始的fastq序列文件mapping 到参考基因组上,生成 sam 格式文件(个人理解比对就是知道每条reads在参考基因组的具体位置,即在哪一条染色体的哪个位置)。下图是使用IGV查看比对的结果,图中看到的这些峰就是我们常说的peaks,最后一组是对照组。
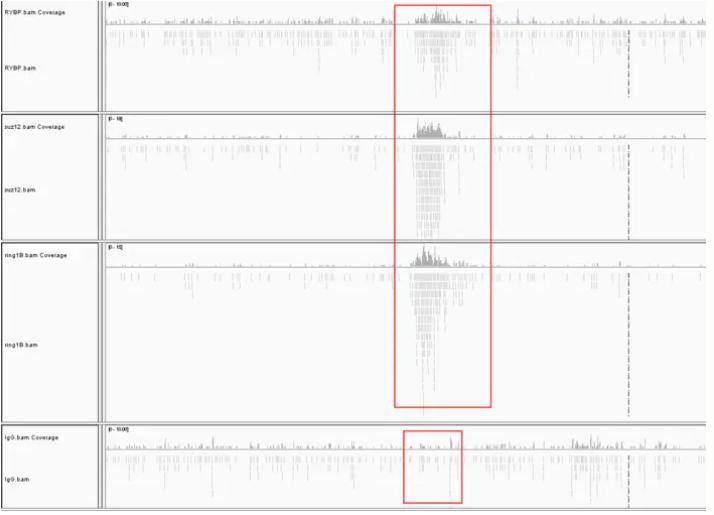
IGV查看比对结果
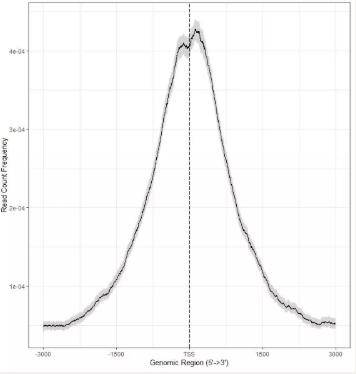
(3) peak calling:寻找peak出现的位置,而这些位置可能就是我们感兴趣的靶蛋白结合的位点。以下两张图显示的是ChIP peak在TSS位点上的富集情况。
下图中的0点即为TSS位点,横坐标表示的是基因组的位置(TSS位点上下游3000bp的范围)。红色表示富集程度。可以看到这些peaks在TSS位点上显著富集。
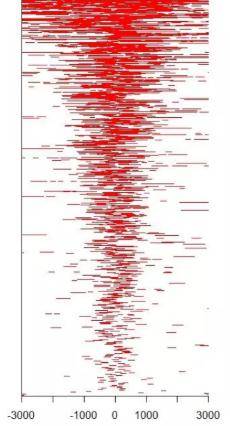
下图携带的信息和上图是一致的(网上找的图片,像素不佳,望见谅)。横轴依然是基因组的位置,中间表示TSS位点,纵轴表示peaks的数目。可以看到peaks也是在TSS位点上显著富集的。
(4) peaks注释:个人理解为寻找ChIP peaks所对应的靶基因。
(5) 下游分析:pathway富集分析、寻找motif、结合其他组学数据.
DNase-seq
原理:DNase-seq使用了限制性内切酶(DNase I)对样品进行了处理。在染色质压缩区域,DNA链被致密结构很好地保护起来,使得内切酶无法接近,只能切割开放区域的DNA。同样的,在开放区域,缠绕在核小体上的DNA被核小体结构所保护,只有核小体之间的DNA序列能够被DNase I切割,这些区域内能够被DNase切割的位点也被称为DHS,即DNase超敏感位点。
MNase-seq
这种测序方法和DNase-seq原理类似,但是探测的区域是互补的。因为MNase-seq使用的酶是限制性外切酶,将不受保护的区域统统切除,只留下核小体上缠绕的DNA序列。
以上两种方法把切割完的DNA拿来测序,和已知的全基因组序列相比较,就能发现被切掉的是哪些地方,没有被切掉的地方又在哪里,从而获知开放的染色质区域。但这两种方法都需要限制性酶,具有序列的特异性,所以缺点是切割下来的片段都不是完整的开放染色质信息。
ATAC-seq
我们需要先理解一个概念:转座是指一段DNA片段从DNA 的一个位点移到另一个位点。通过转座酶来实现,也是需要插入位点的染色质是开放的。
原理:ATAC-seq使用改造的Tn5转座酶,将转座DNA(已知DNA序列)设计为接头,随机插入染色质的开放区域。开放染色质都具有转座酶敏感性,所以他可以得到完整的开放染色质的区域。再利用已知序列的标签进行PCR后送去测序,就知道哪些区域是开放染色质了。ATAC-seq中的峰,往往是启动子、增强子序列,以及一些反式调控因子结合的位点。
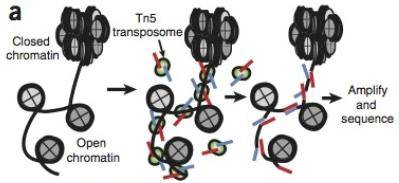
ATAC-seq有一个特点:两个接头置换出来的有可能是开放性染色质的区域,也有可能是转录因子上的DNA序列。这一点从上图中就可以看出来。所以在ATAC-seq的峰中,既有对应开放性染色质的,也有对应核小体的DNA片段上的。
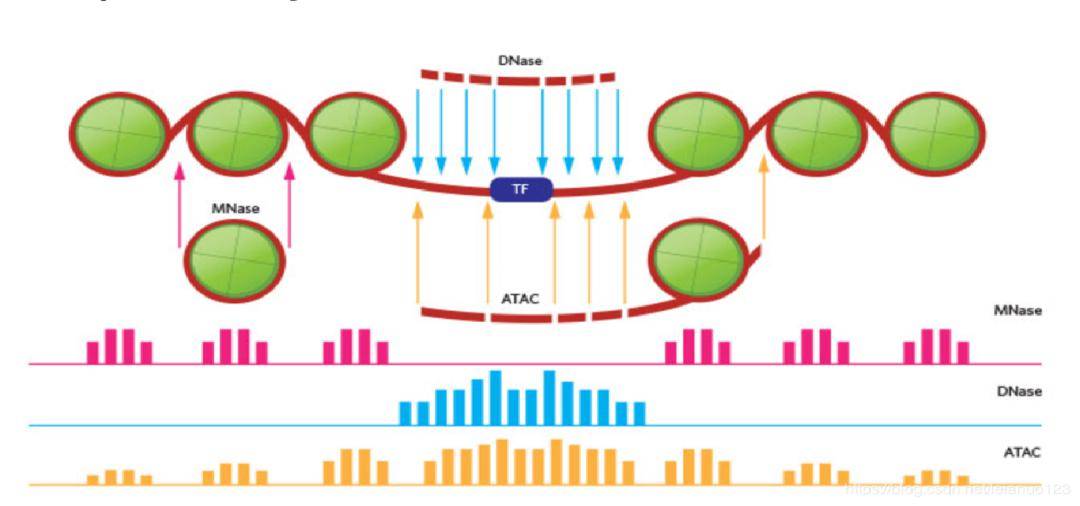
ATAC-seq的实验流程:裂解细胞获得细胞核→使用Tn5转座酶酶切并纯化,最后回收DNA片段→PCR扩增→测序。
ATAC-seq的分析流程:
1、数据预处理
(1)比对前质量控制:FastQC可用于在测序数据中可视化碱基质量得分、GC含量、序列长度分布等。
(2)原始序列比对:将过滤的read比对到参考基因组。
(3)比对后处理和质量控制:
比对后处理就是去除重复序列和细胞器序列。
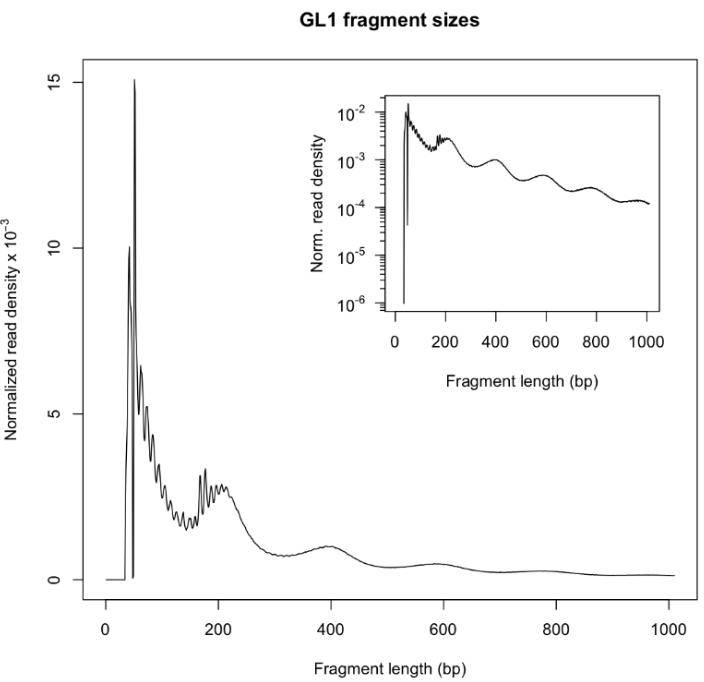
通常,成功的ATAC-seq实验应生成片段大小分布图(从bam文件得到),其具有递减的和周期性的峰,对应于无核小体区域(NFR)(<100 bp)和单核、双核和三核小体(~200, 400,600碱基对)。因为大多数的Linker DNA的大小介于10-80bp之间,所有得到的大多数片段都会是小于100bp的(前面那段毛毛刺刺的,密度又很高的)。而每个Nucleosome的DNA大小为180bp左右,加上两边插入进的冗余,我们会得到大约200bp长度是mono-nucleosome的DNA。如果是两个Nucleosome之间的片段的话,就是400bp左右。依此类推。如下图是典型的ATAC-seq片段大小分布图。
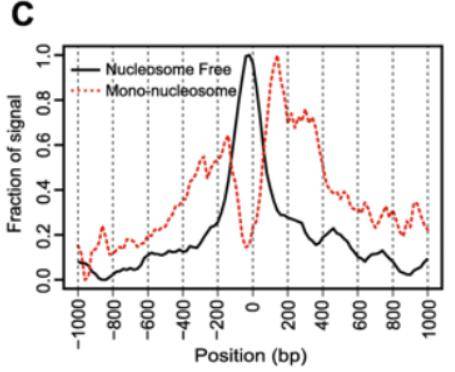
无核小体区域的片段应该在基因的转录起始位点(TSS)周围富集,而核小体结合区域的片段应该在TSS处被形成低谷,TSS周围的侧翼区域会稍微富集。可以使用工具ATACseqQC进行评估。
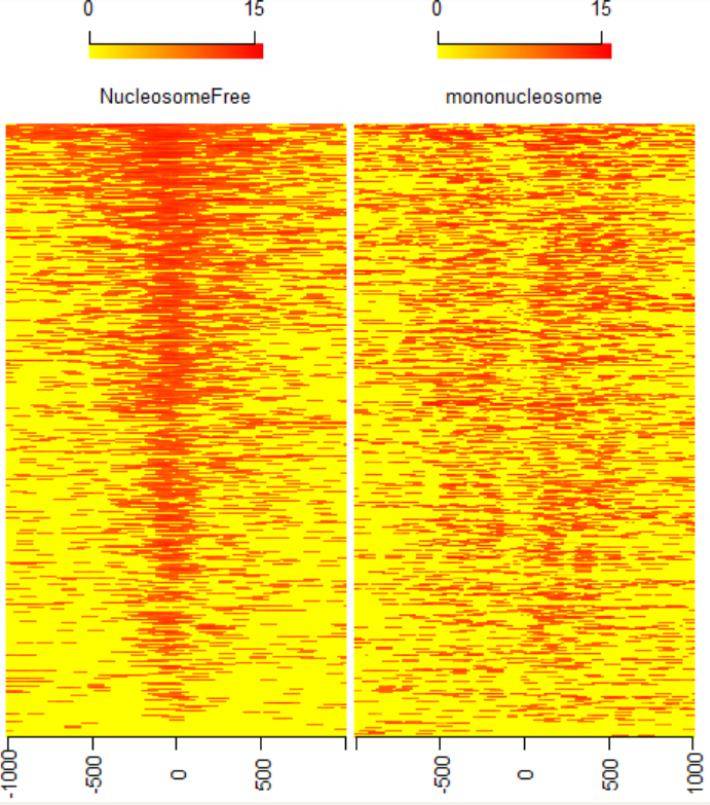
TSS附近信号的分布也可以用热图来显示。
2、Peak-calling:也就是从比对得到BAM文件中找出reads的覆盖区,也就是那个峰出现的位置。
3、高级分析
(1)peak差异分析:寻找不同分组之间的差异peaks
(2)peak注释:峰的注释可将染色质的可及性与基因调控联系起来。通常,峰会被注释到最接近的基因或调控元件。获得最接近的基因之类的基因列表后,还可以使用GO,KEGG和Reactome等数据库进行功能富集分析。
(3)motif富集分析:得到每个peak region里motif的位置和频率,再和随机背景或其它条件比较,就可以做motif的富集分析。
(4)footprint分析:ATAC-seq中的footprint是指一个TF结合在DNA上,阻止Tn5切割,在染色质开放区域留下一个相对缺失的位置。而TF周围的组蛋白因为TF造成的空间的推挤反而形成了开放度比较高的区域。
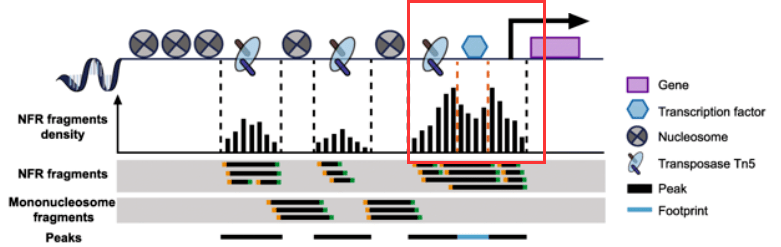
(5) 核小体定位分析:分析核小体的结合区域。
ATAC-seq数据与多组学数据整合:
- ATAC测序+转录组数据
通过联合转录组测序结果,看ATAC上测到的一些开放性高的DNA序列区域,是否对应的转录本表达量也有增加,也可以找到对应的转录本相关基因的上游调控序列,从而从整体上分析从DNA到RNA的转录过程。进一步对基因进行功能分析,再结合实验表型进行讨论,我们就可以理清楚表观调控-表达-功能-表型这样一个过程的相关机制。
- ATAC测序+ChIP-seq
ATAC测序≠ChIP-seq。ATAC测序之后也需要做ChIP-seq来做进一步的验证,通过ChIP的测序结果,来进一步对ATAC所预测到的一些转录因子结合区域是否真的有转录因子结合进行验证。
- ATAC测序+甲基化数据
已证明在整个基因组上核小体的定位影响DNA的甲基化状态,从同一个DNA分子得到DNA甲基化和核小体的定位可以使我们能够更好地了解DNA甲基化和核小体定位是如何共同控制染色质的可接近性。
参考资料:
https://www.plob.org/article/18583.html
https://www.jianshu.com/p/68f99d4bcb7b
https://mp.weixin.qq.com/s/vgXrscsUkfYdC5qt_PUNlw
https://www.jianshu.com/p/a7b6ce208f98?utm_campaign=haruki&utm_content=note&utm_medium=reader_share&utm_source=weixin
https://www.jianshu.com/p/2b8e2ea26665
https://www.jianshu.com/p/32b2fab75c24
http://www.biomarker.com.cn/archives/13555
http://qiubio.com/new/book/
- 本文固定链接: https://maimengkong.com/kyjc/1196.html
- 转载请注明: : 萌小白 2022年10月1日 于 卖萌控的博客 发表
- 百度已收录
