宏基因组/宏转录组测序
2022-4-3 萌小白
宏基因组学这一概念最早是在1998年由威斯康辛大学植物病理学部门的Jo
Handelsman等提出的,是源于将来自环境中基因集可以在某种程度上当成一个单个基因组研究分析的想法,而宏的英文是“meta-”,具有更高层组织结构和动态变化的含义。后来加州伯克利分校的Kevin
Chen和Lior
Pachter将宏基因组定义为“应用现代基因组学的技术直接研究自然状态下的微生物的有机群落,而不需要在实验室中分离单一的菌株”的科学。
宏基因组学研究的对象是特定环境中的总DNA,不是某特定的微生物或其细胞中的总DNA,不需要对微生物进行分离培养和纯化,这对我们认识和利用未培养微生物提供了一条新的途径。
宏基因组测序无需PCR扩增,其实验流程见下图:
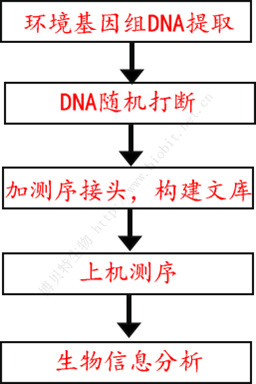
图1 宏基因组/宏转录组实验流程
宏基因组数据的标准分析流程通常包括质量控制、rRNA识别、序列组装 (assembly)、基因预测 (gene calling)、基因注释
(功能和生物来源的annotation)
等步骤(如下图)。其个性化分析多种多样,例如多样性分析(PCoA、NMDS等排序分析、PerMANOVA/Anosim分析)、LEfSE分析、与环境因子的关联分析
(Mantel test、方差分解分析VPA等)、网络分析,等等。
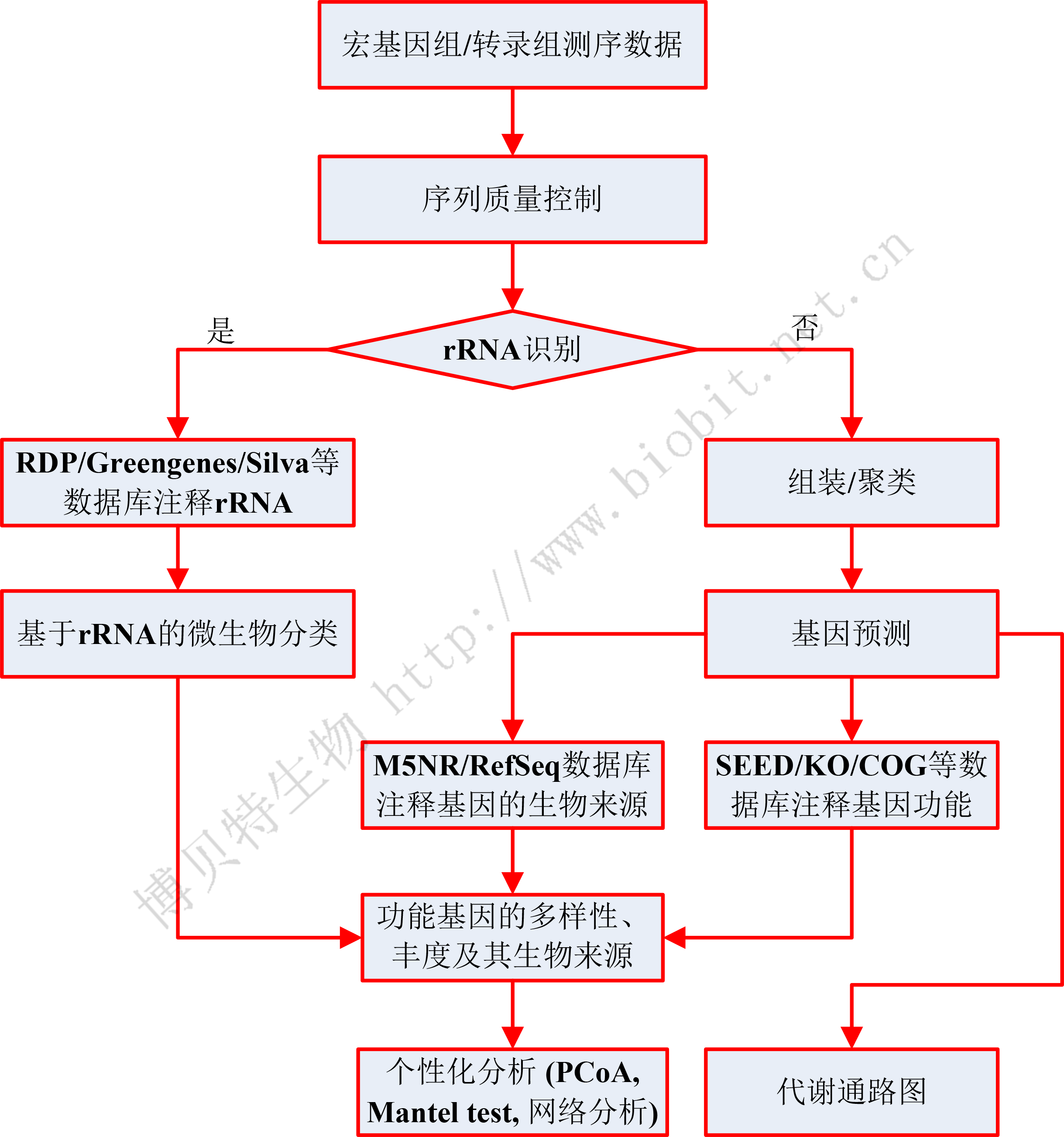
图2 宏基因组/宏转录组生物信息分析流程
宏基因组生物信息分析结果示例如下:
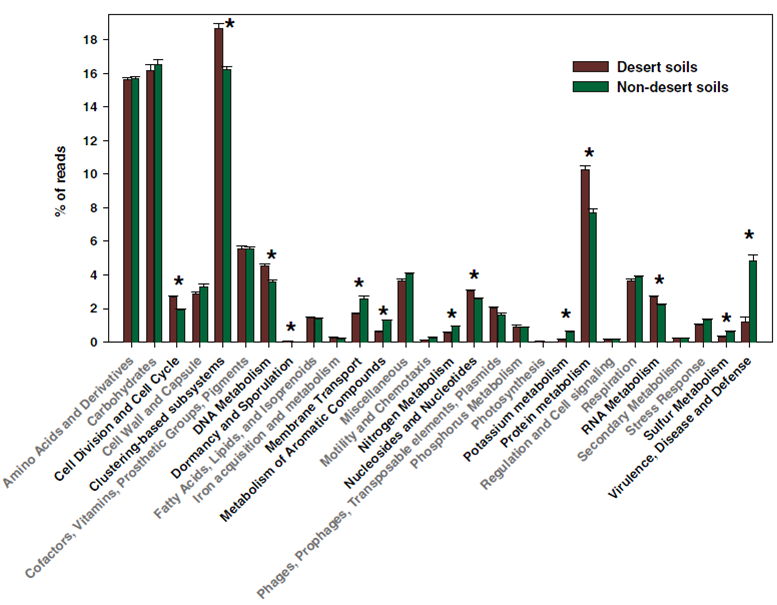
图3 用柱状图呈现不同处理之间功能类别(SEED subsystems功能库Level 1 水平)的丰度差异(Fierer et al. PNAS 2012)
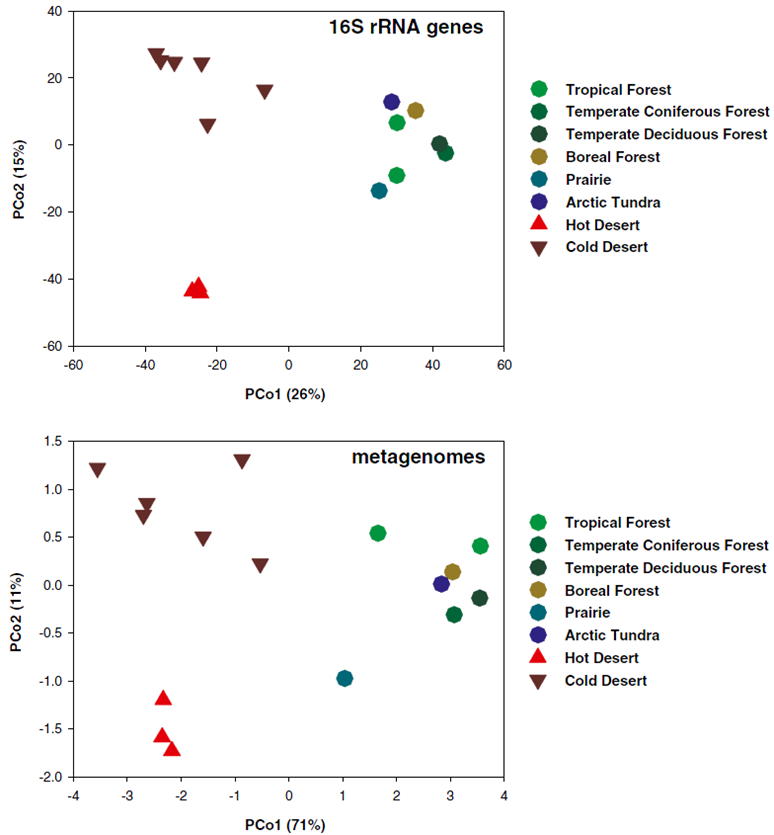
图4 用PCoA图呈现微生物群落(a. 16S rRNA)与功能(b. 宏基因组)在beta多样性上的差异(Fierer et al. PNAS 2012)
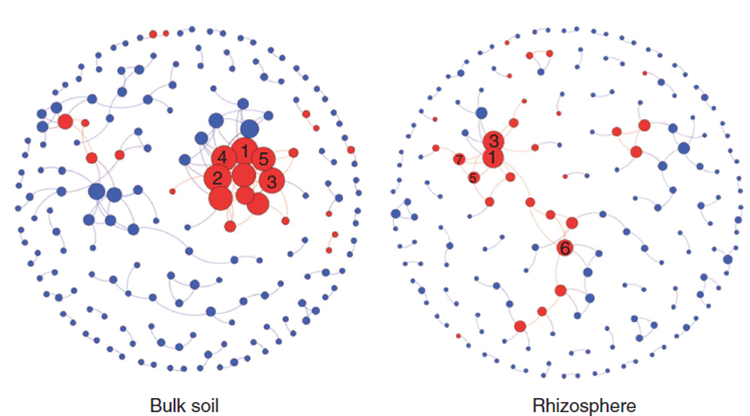
图5 比较不同处理(非根际土与根际土)之间的微生物功能网络(Menders et al. ISME J 2014)
图中蓝色节点表示功能,红色节点为细菌门,连线表示两节点之间存在显著相关。
Handelsman等提出的,是源于将来自环境中基因集可以在某种程度上当成一个单个基因组研究分析的想法,而宏的英文是“meta-”,具有更高层组织结构和动态变化的含义。后来加州伯克利分校的Kevin
Chen和Lior
Pachter将宏基因组定义为“应用现代基因组学的技术直接研究自然状态下的微生物的有机群落,而不需要在实验室中分离单一的菌株”的科学。
宏基因组学研究的对象是特定环境中的总DNA,不是某特定的微生物或其细胞中的总DNA,不需要对微生物进行分离培养和纯化,这对我们认识和利用未培养微生物提供了一条新的途径。
宏基因组测序无需PCR扩增,其实验流程见下图:
图1 宏基因组/宏转录组实验流程
宏基因组数据的标准分析流程通常包括质量控制、rRNA识别、序列组装 (assembly)、基因预测 (gene calling)、基因注释
(功能和生物来源的annotation)
等步骤(如下图)。其个性化分析多种多样,例如多样性分析(PCoA、NMDS等排序分析、PerMANOVA/Anosim分析)、LEfSE分析、与环境因子的关联分析
(Mantel test、方差分解分析VPA等)、网络分析,等等。
图2 宏基因组/宏转录组生物信息分析流程
宏基因组生物信息分析结果示例如下:
图3 用柱状图呈现不同处理之间功能类别(SEED subsystems功能库Level 1 水平)的丰度差异(Fierer et al. PNAS 2012)
图4 用PCoA图呈现微生物群落(a. 16S rRNA)与功能(b. 宏基因组)在beta多样性上的差异(Fierer et al. PNAS 2012)
图5 比较不同处理(非根际土与根际土)之间的微生物功能网络(Menders et al. ISME J 2014)
图中蓝色节点表示功能,红色节点为细菌门,连线表示两节点之间存在显著相关。
发表评论: