研究表明,几乎所有生物的表型变异,环境适应和物种形成都与基因组间的结构变异有关。人类基因组既包括蛋白质编码基因,也包括控制这些基因何时表达以及表达到何种程度的调控信息。结构变异(Structure
Variantions,简称SVs)是造成物种表型差异的一个重要原因,且与各类疾病,特别是癌症的发生、发展紧密相关,因此研究结构变异非常重要。基因组结构变异通常是指长度大于1Kb的基因组序列变异,包括多种不同的类型:插入(insertion)、缺失(deletion)、反转(inversion)、异位(translocation)、拷贝数变异(copy
number variation,CNV或者duplication)。
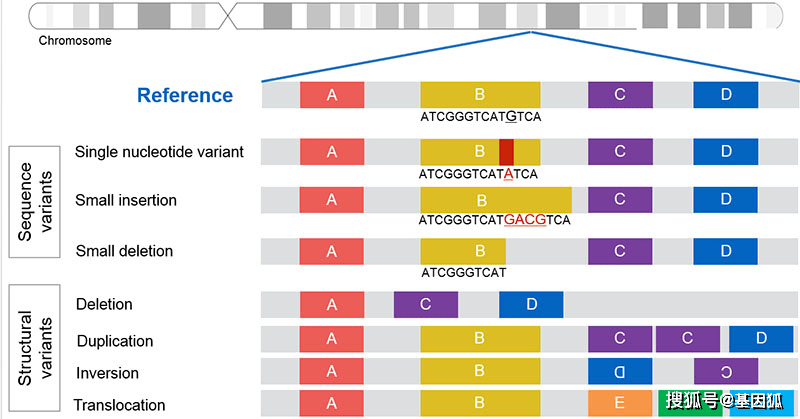 不同类型的基因组变异示意图(图片来源:labspaces)
不同类型的基因组变异示意图(图片来源:labspaces)
有研究发现,基因组上的SVs比起SNP而言,更能代表人类群体的多样性特征。SVs对基因组的影响比起SNP更大,一旦发生往往会给生命体带来重大影响,比如导致出生缺陷、癌症等。近日,一个隶属于美国各地大量医疗机构的大型研究团队,以及三个来自芬兰、两个来自墨西哥和一个来自英国的研究人员,利用17795个已经深度测序的基因组序列绘制了目前最新的结构变异图谱,项研究成果发表在近期的《自然》杂志上。
 论文截图
论文截图
研究团队他们使用了可扩展的流程对17,795个深度测序的人类基因组中的SV进行了分析。平均每个个体携带2.9个稀有SV,这些SV会改变编码区,影响4.2个基因的剂量或结构,占稀有高影响力编码等位基因的4.0-11.2%。根据计算模型,他们估计SVs占全基因组罕见等位基因的17.2%,其预测的有害影响与功能丧失编码的等位基因相当。大约90%的此类SV是非编码缺失(每个基因组平均19.1)。他们报告了158,991个超稀有SV,并显示大约2%的人携带超稀有的兆碱基规模SV,其中近一半是平衡或复杂的重排。
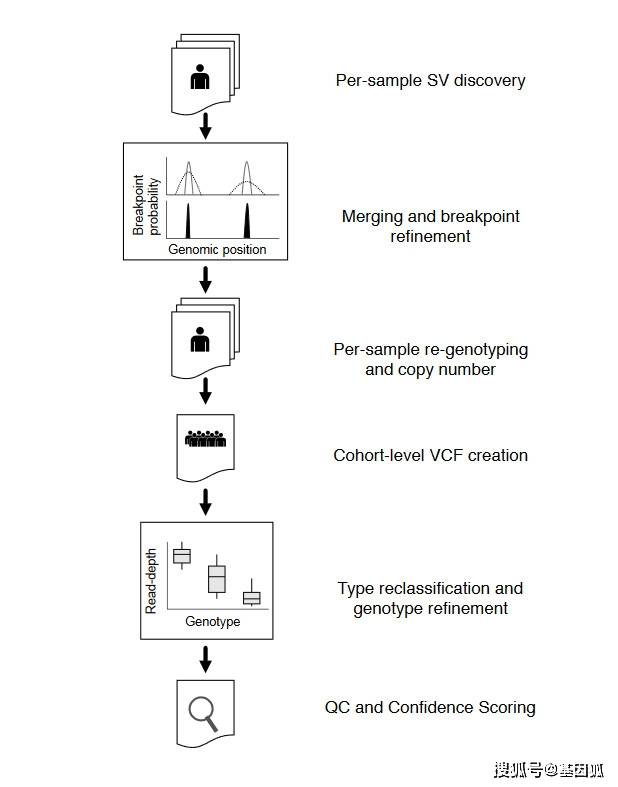 Callset construction pipeline.
Callset construction pipeline.
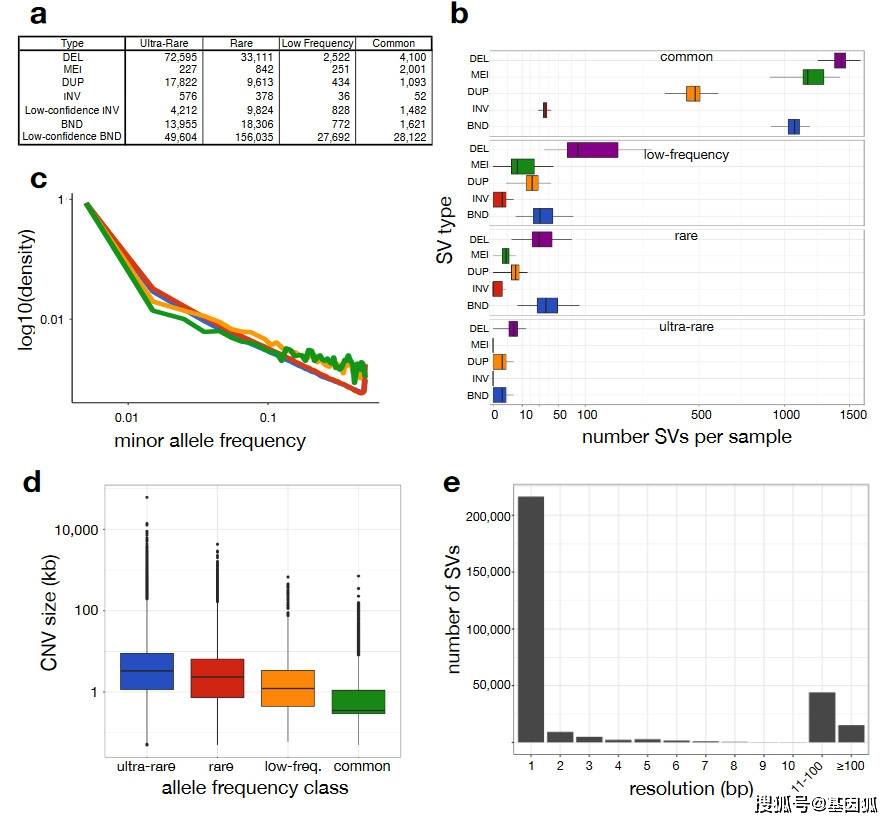 Version of the “B38” callset derived from 14,623 samples
Version of the “B38” callset derived from 14,623 samples
(a)Number of high-confidence and low-confidence SVs by class and
frequency bin. SV classes are defined as: DEL, deletion; MEI, mobile
element insertion; DUP, duplication; INV, inversion; BND, “break-end”,
which is a generic term in the VCF specification for SV breakpoints that
cannot be unequivocally classified. Minor allele frequency (MAF) bins
are defined as: “ultra-rare” is private to an individual or family;
“rare” is MAF<1%; “low-frequency” is 1%<MAF<5%; “common” is
MAF>5%.
(b)Number of SVs per sample (x-axis, square-root scaled) by SV type (y-axis) and frequency class (panels labelled at top).
(c)MAF distribution for SNV, indel, deletion (DEL) and duplication
(DUP) variants for a subset of 4,298 samples for which GATK-based
SNV/indel were also available.
(d)CNV length distributions for each frequency class, defined as in
part (a). (e)Histogram showing the resolution of SV breakpoint calls, as
defined by the length of the 95% confidence interval of the
breakpoint-containing region defined by LUMPY, after cross-sample
merging and refinement using svtools.
最后,他们推断基因和非编码元件的剂量敏感性,揭示与元件类别和保守性有关的趋势。这项工作将有助于指导WGS时代的SV分析和注释。
Abstract:
A key goal of whole-genome sequencing (WGS) for human genetics
studies is to interrogate all forms of variation, including single
nucleotide variants (SNV), small insertion/deletion (indel) variants and
structural variants (SV). However, tools and resources for the study of
SV have lagged behind those for smaller variants. Here, we used a
scalable pipeline22 to map and characterize SV in 17,795 deeply
sequenced human genomes. We publicly release site-frequency data to
create the largest WGS-based SV resource to date. On average,
individuals carry 2.9 rare SVs that alter coding regions, affecting the
dosage or structure of 4.2 genes and accounting for 4.0-11.2% of rare
high-impact coding alleles. Based on a computational model, we estimate
that SVs account for 17.2% of rare alleles genome-wide with predicted
deleterious effects equivalent to loss-of-function coding alleles;
approximately 90% of such SVs are non-coding deletions (mean 19.1 per
genome). We report 158,991 ultra-rare SVs and show that around 2% of
individuals carry ultra-rare megabase-scale SVs, nearly half of which
are balanced or complex rearrangements. Finally, we infer the dosage
sensitivity of genes and non-coding elements, revealing trends related
to element class and conservation. This work will help guide SV analysis
and interpretation in the era of WGS.
DOI: 10.1038/s41586-020-2371-0