表达谱分析中,经常会使用到相关性分析,探索一组基因间的共表达特征。例如,这些基因间的表达是否存在较强的协同性,一个基因表达值的改变是否与另一个基因表达值改变显著相关,它们之间是共激活还是抑制关系等。
对于相关性分析结果的可视化,通常有多种方法,例如相关性散点图、弦图、共表达网络图。本篇教程则主要带大家了解如何基于基因表达值的相关性,绘制弦图。
如下弦图来自文献“CUL4B promotes prostate cancer progression by forming positive feedback loop with SOX4”,共涉及了7个基因。图中的连线就代表了基因间表达值的相关性信息,红色代表正相关,绿色代表负相关,颜色越深或连线越粗代表相关强度越高。从图中可以看出,这几个基因间以较强的正相关关系占主导,暗示它们之间的共激活关系,或者在相似的生物学过程中发挥作用。
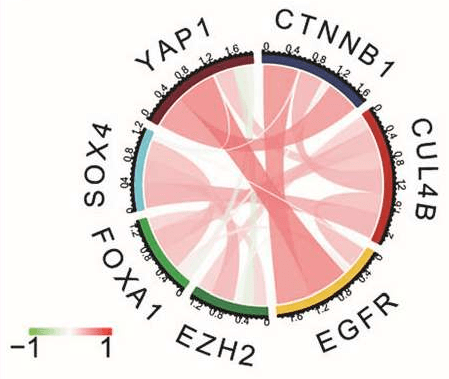 文献中的弦图,表示基因间表达值的相关性
文献中的弦图,表示基因间表达值的相关性
接下来,我们模仿文献中的图片样式,绘制基因表达相关性弦图。
作图原始数据、R代码等,可关注微信公众号获取。
1.计算基因表达值的相关性
为了绘制弦图,首先需要计算基因表达值的相关性。
来看提供的示例数据“gene_FPKM.txt”,记录了6个基因在30个样本中的表达值(FPKM值)信息。将该基因表达值矩阵读入到R中,计算基因表达值的Pearson相关性。
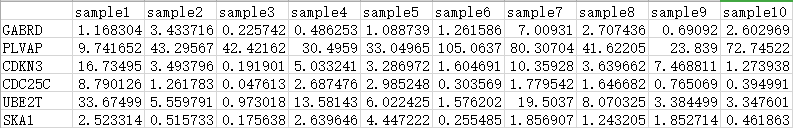 示例数据格式
示例数据格式
!!!**************************************************
#读取基因表达值数据
gene <- read.delim('gene_FPKM.txt', row.names = 1, sep = '\t')
gene <- t(gene) #转置数据,使行为样本,列为基因
#表达值进行log(1+)转化,使数据更服从正态分布,减少离散度极大值影响
gene <- log(gene+1)
#基因表达值的相关性分析,以Pearson相关系数为例
gene_cor <- cor(gene, method = 'pearson')
#去除基因的自相关,也就是对角线的值
diag(gene_cor) <- 0
gene_cor #最终的基因间表达值Pearson相关性矩阵
!!!**************************************************
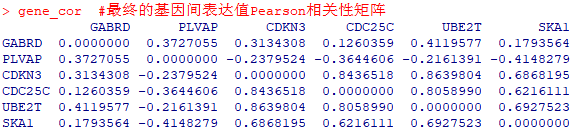 计算基因间表达值的相关性
计算基因间表达值的相关性
初步计算了6个基因间表达值的Pearson相关系数,正值代表正相关,负值代表负相关,绝对值越大代表相关强度越高。
2.绘制相关性弦图
再对上述得到的相关性矩阵做个转换,得到一种两两对应的结构,如下示例。
!!!**************************************************
#将获得的相关性矩阵转换为两两对应的数据框结构
gene_cor <- reshape2::melt(gene_cor)
gene_cor <- subset(gene_cor, value != 0) #去除0值的相关性
head(gene_cor) #前两列是两个基因名称,第三列为两个基因的相关性
!!!**************************************************
 基因间两两相关性
基因间两两相关性
最后,可通过circlize包中的方法绘制弦图,circlize包是R语言中绘制弦图的一款非常优秀的R包。
!!!**************************************************
#绘制弦图
library(circlize)
chordDiagram(gene_cor,
annotationTrack = c('grid', 'name', 'axis'), #绘制外周圆弧区,显示名称和刻度轴
grid.col = c(GABRD = 'green3', PLVAP = 'red', CDKN3 = 'orange', CDC25C = 'purple', UBE2T = 'skyblue', SKA1 = 'blue'), #定义基因颜色
col = colorRamp2(c(-1, 0, 1), c('green', 'white', 'red'), transparency = 0.5), #根据相关性大小展示连线的颜色范围
annotationTrackHeight = c(0.05, 0.05), #名称离圆弧的距离,以及圆弧的宽度
!!!**************************************************
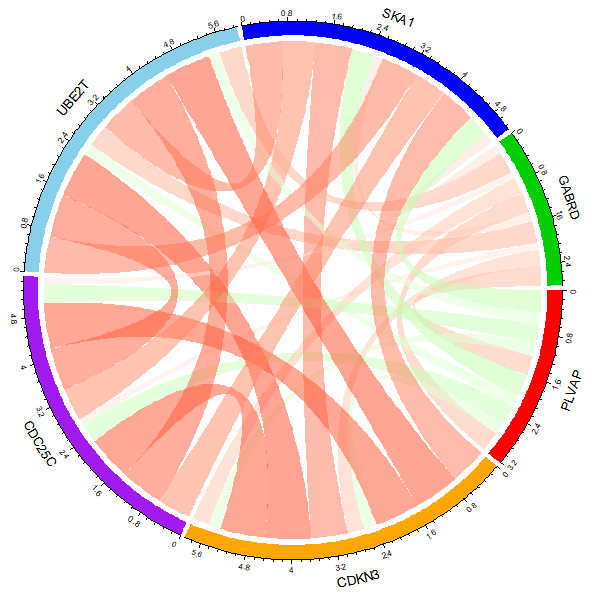 R包circlize绘制的弦图
R包circlize绘制的弦图
这样弦图就得到了,连线表示了6个基因间表达值的Pearson相关性信息,红色代表正相关,绿色代表负相关,颜色越深或连线越粗代表相关强度越高。
!
!
!
注:!!!*******之间为R脚本内容