经典功能富集分析可戳:
《KEGG富集分析及可视化,一把子拿捏!》
《GO富集分析及可视化,一把子拿捏!》
通过KEGG、GO等经典富集分析,我们能够了解到阈值筛选出的差异基因参与的通路和具备的功能,以及哪些功能/通路对表型变化是可能起主导作用的。
但在经典富集分析中,我们无法得知某条通路下差异基因的总体变化趋势,即富集到同一通路下的基因既又上调也有下调,那么这条通路的表现形式到底是被激活了呢?还是被抑制了呢?且设定的阈值可能也会卡掉一些在统计学意义上无显著差异但实则有着重要生物学意义的基因,导致重要通路被遗漏。
上述的问题我们可以采用GSEA富集分析解决。GSEA针对所有基因而非仅差异基因,可以检测出不显著但是一致的差异表达趋势的基因集,同时,还能够判断通路处于被激活还是抑制状态。
本期学习内容为使用clusterProfiler及系列R包完成GSEA富集及可视化,包括对不同数据库如KEGG、GO、Reactome、Do、MSigDB进行GSEA富集。
#GSEA富集分析
#部分所需R包载入:
library(org.Hs.eg.db)#物种注释包(Homo sapiens)
library(clusterProfiler)#富集分析
#DESeq2差异分析结果表格导入:
load( "TCGA_CHOL_DESeq2.Rdata")
head(DESeq2)
length(rownames(DESeq2))#共16345个基因
#添加entrez ID列:
##:symbol转entrez ID:
symbol<- rownames(DESeq2)
entrez<- bitr(symbol,
fromType= "SYMBOL",#现有的ID类型
toType= "ENTREZID",#需转换的ID类型
OrgDb= "org.Hs.eg.db")
head(entrez)
#准备genelist文件:
##需要的genelist格式:entrez ID+log2fc
genelist<- DESeq2$log2FoldChange
names(genelist) <- rownames(DESeq2)
head(genelist)
#genelist过滤(ID转换中丢失的部分基因):
genelist<- genelist[names(genelist) %in% entrez[,1]]
names(genelist) <- entrez[match(names(genelist),entrez[,1]),2]
length(genelist)#过滤后剩16161个基因
head(genelist)
#将genelist按照log2FC值从高到低进行排序:
genelist<- sort(genelist,decreasing = T)
head(genelist)
#GSEA_KEGG富集分析:
KEGG_ges<- gseKEGG(
geneList= genelist,
organism= "hsa",#不同物种选择可官方查询:https://www.genome.jp/kegg/catalog/org_list.html
minGSSize= 10,
maxGSSize= 500,
pvalueCutoff= 0.05,
pAdjustMethod= "BH",
verbose= FALSE,
eps= 0
)
#提取结果表:
KEGG_ges_result<- KEGG_ges@result
#显示前10列(第十一列为富集到该基因集的核心基因列表,这里过长不展示):
KEGG_ges_result[1:3,1:10]
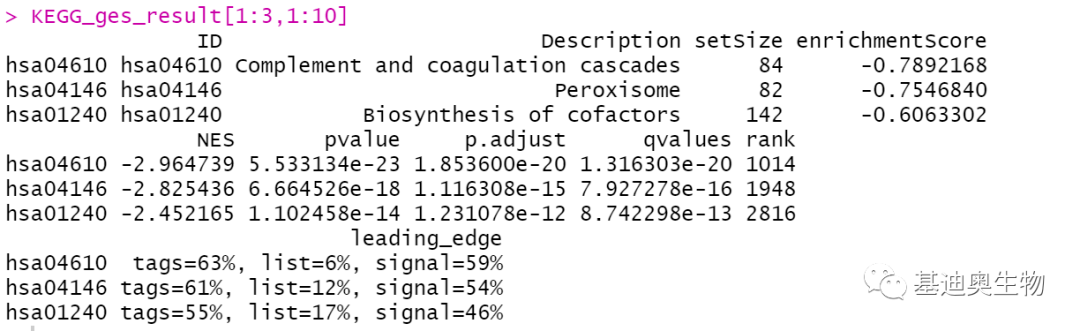
我们简单介绍下GSEA富集结果中不同列的含义:
ID:KEGG pathway的 ID名(对应所使用的数据库通路/功能基因集的ID)
Deion:基因集(pathway)描述
setSize:富集到该pathway的基因数
enrichmentScore:富集分数(ES),即GSEA的核心
NES:归一化后的Enrichment score(ES)
pvalue/p.adjust/qvalues:p值/矫正后p值/FDR值
rank:在基因集中对ES分数贡献最大的核心基因在genelist排序中的位置(按照log2fc从大到小的排序)
leading_edge:三个指标的含义(如下)
tag:核心基因在该基因集基因总数的占比
list:核心基因在所有基因总数的占比
signal:由tag和list指标计算。对于一个基因集而言,当核心基因的数目和该基因集下的基因总数相同时,signal取值最大,当该基因集的基因数目和所有基因数目接近时,signal的取值接近于0。
core_enrichment:富集到该基因集(pathway)的基因列表
ES/NES大于0的基因集(pathway),我们认为该通路被激活或者说是高表达的,小于0认为被抑制或者低表达。
#GSEA可视化:
##山峦图:
library(ggridges)
library(ggplot2)
library(enrichplot)
p<- ridgeplot(KEGG_ges,
showCategory= 15,
fill= "p.adjust",
decreasing= T)
p

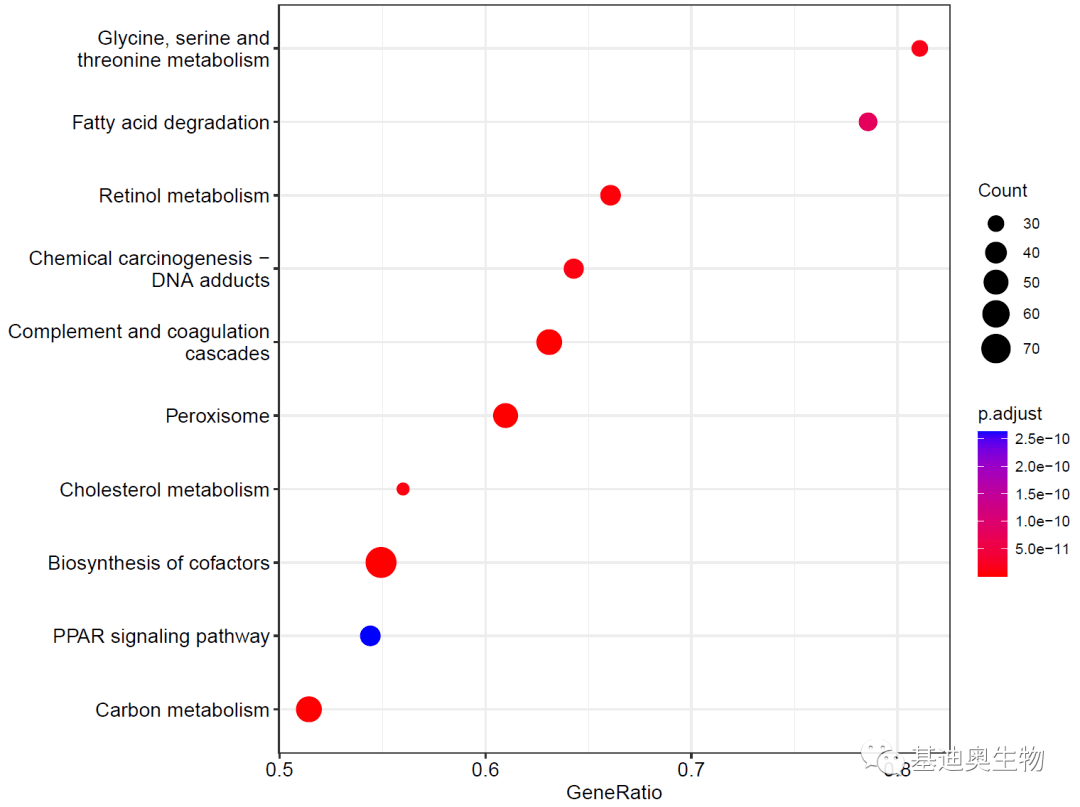
##气泡图:
pp<- dotplot(KEGG_ges)
pp
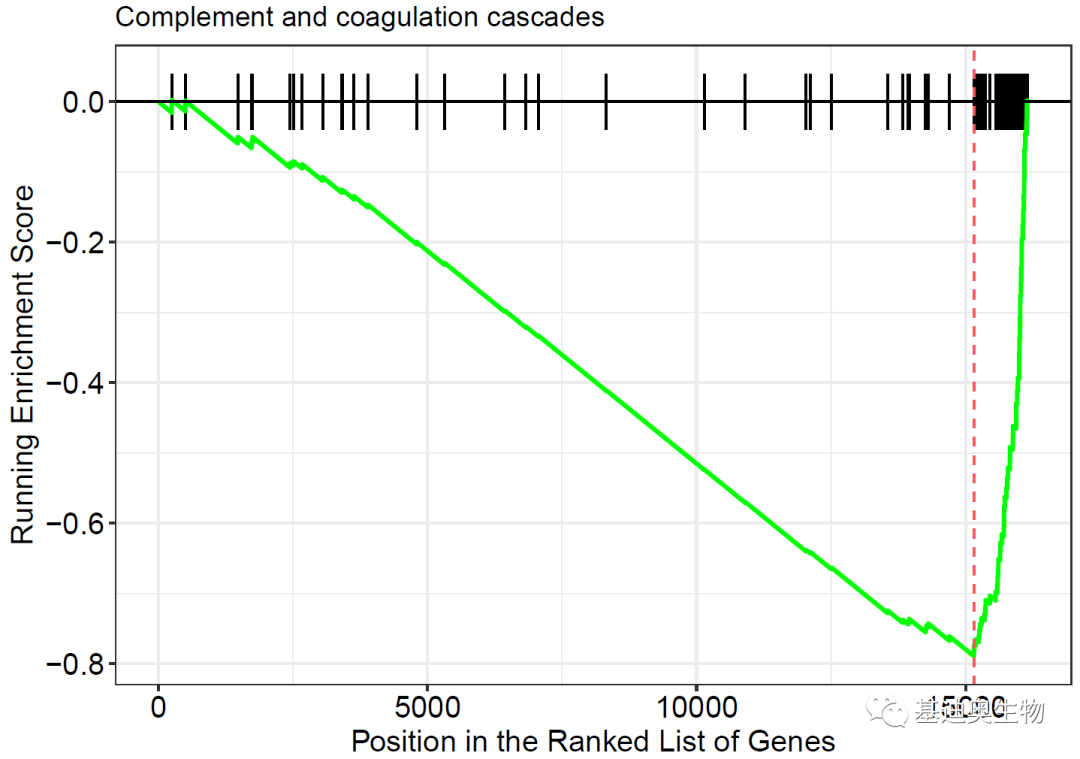
#gseaplot函数:
##enrichmentScore折线图:
p1<- gseaplot(KEGG_ges,
geneSetID= 1, #取第一个pathway绘图
by= "runningScore", #or “preranked”, “ all”
title= KEGG_ges$Deion[1])
p1
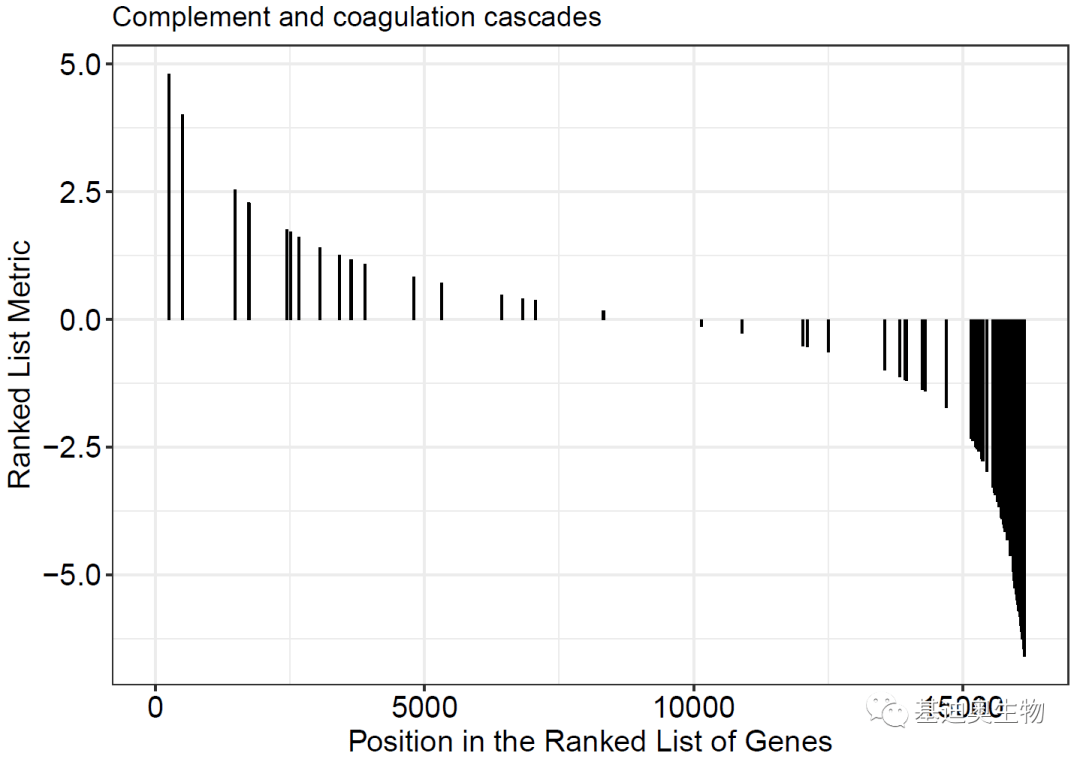
##rank分布图:
p2<- gseaplot(KEGG_ges,
geneSetID= 1,
by= "preranked",
title= KEGG_ges$Deion[1])
p2
##同时显示:
p3<- gseaplot(KEGG_ges,
geneSetID= 35,
by= "all",
title= KEGG_ges$Deion[35])
p3
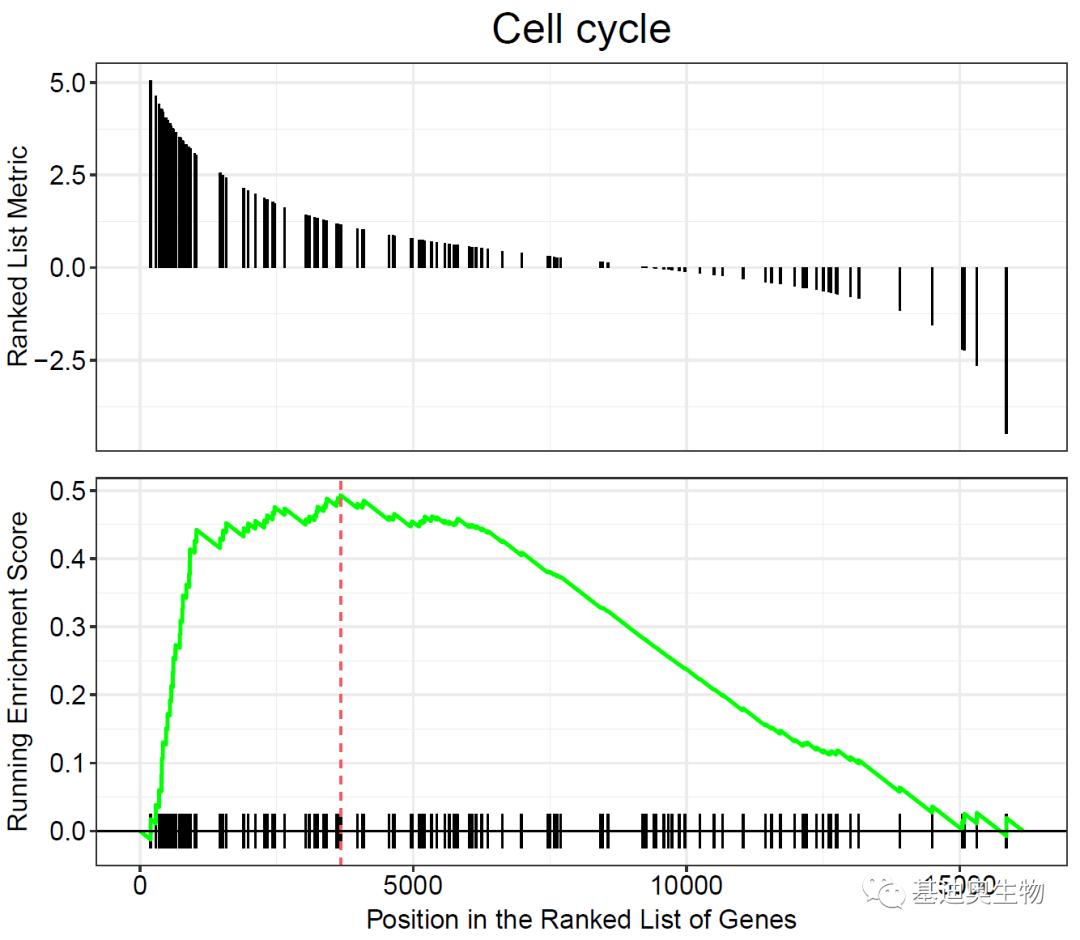
使用gseaplot绘制出的效果一般,所以我们一般会使用gseaplot2函数进行绘制,还可以将多个基因集的ES折线合并在同一张图表中。
#gseaplot2函数:
##单个基因集展示:
p4<- gseaplot2(KEGG_ges,
geneSetID= 48,
color= "red",
rel_heights= c(1.5, 0.5, 1),#子图高度
subplots= 1:3,#显示哪些子图
pvalue_table= F,#是否显示pvalue表
title= KEGG_ges$Deion[48],
ES_geom= "line")# "dot"将线转换为点
p4
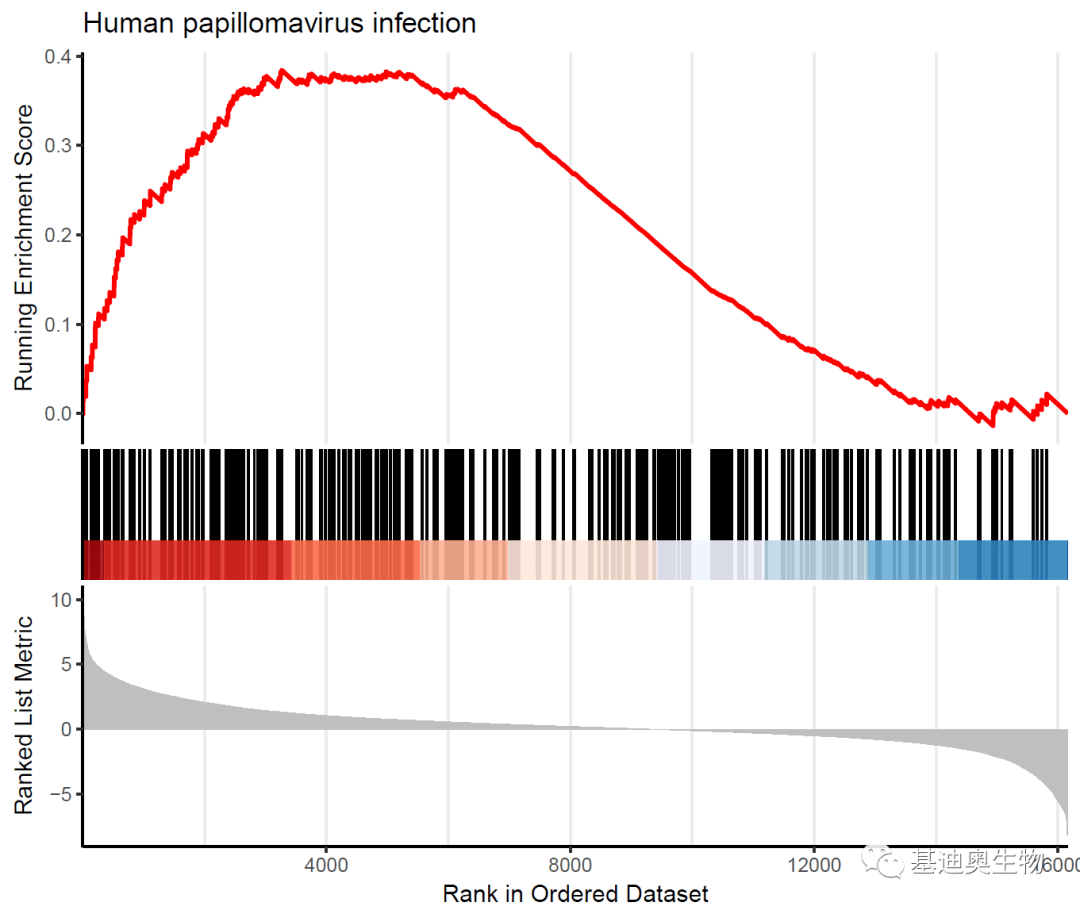
#同时展示多个基因集:
p5<- gseaplot2(KEGG_ges,
geneSetID= 47:49,#或直接输入基因集ID向量名,如c( "hsa04610", "hsa00260")
color= c( "#003399", "#FFFF00", "#FF6600"),
pvalue_table= TRUE,
ES_geom= "line")# "dot"将线转换为点
p5
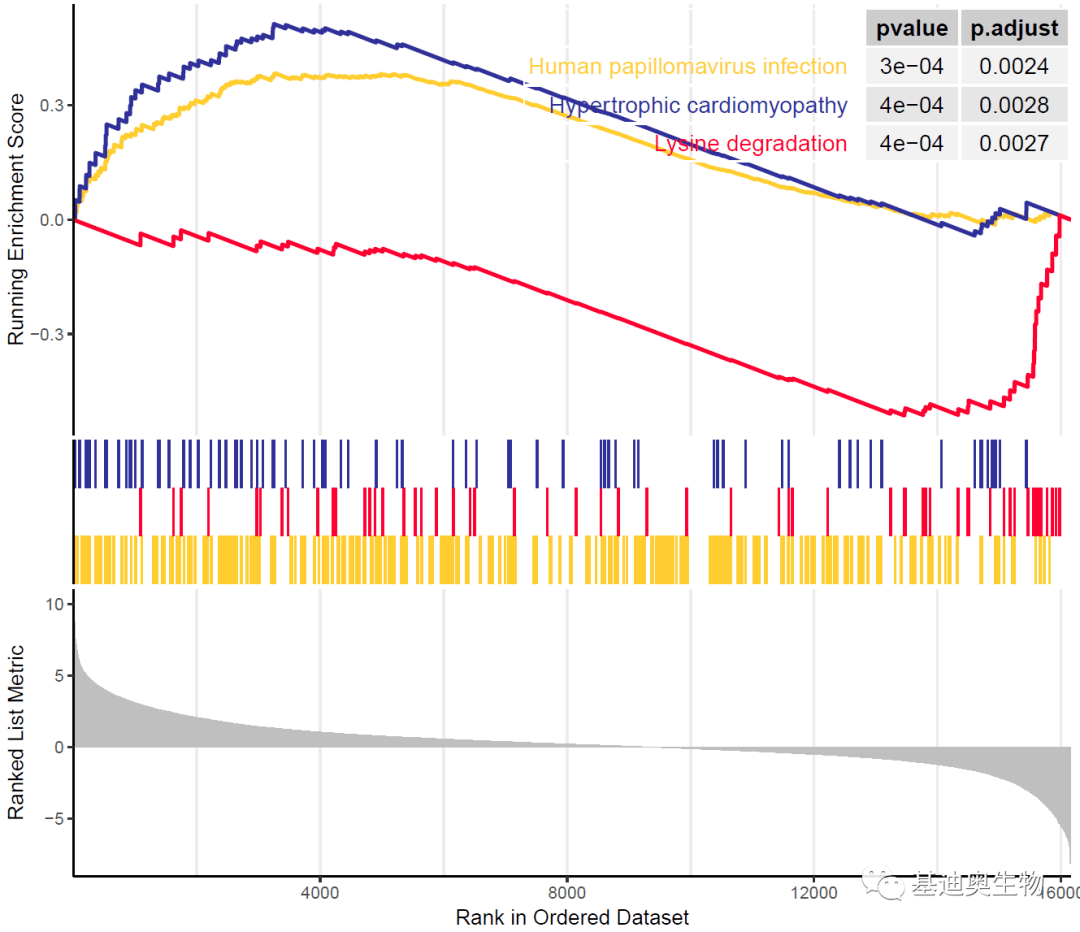
除了KEGG,clusterProfiler包也可以用来完成GO、Reactome数据库的GSEA富集:
##GSEA_GO富集分析:
GO_ges<- gseGO(geneList = genelist,
OrgDb= org.Hs.eg.db,
ont= "CC", #one of "BP", "MF", and "CC"subontologies, or "ALL"for allthree.
minGSSize= 10,
maxGSSize= 500,
pvalueCutoff= 0.05,
eps= 0,
verbose= FALSE)
GO_ges[1:3,1:10] #展示同样省略最后一列

##GSEA_Reactome富集分析:
BiocManager::install( "ReactomePA")
library(ReactomePA)
ret_ges<- gsePathway(genelist,
organism= "human",
minGSSize= 10,
maxGSSize= 500,
pvalueCutoff= 0.05,
pAdjustMethod= "BH",
verbose= FALSE,
eps= 0)
ret_ges[1:3,1:10]

如何想要使用DO(Disease Ontology)数据库进行GSEA,可以使用DOSE包:
##GSEA_DO(Disease Ontology)富集分析:
library(DOSE)
DO_ges<- gseDO(genelist,
minGSSize= 10,
maxGSSize= 500,
pvalueCutoff= 0.05,
pAdjustMethod= "BH",
verbose= FALSE,
eps= 0)
DO_ges[1:3,1:10]

MSigDB(GSEA提供的基因集数据库)现在使用起来也非常方便!可以和msigdf包配合使用,这个包将MSigDB数据用data frame进行了打包,最新数据是一个fork版的(更新到v6.2),我们通过github进行安装。
##GSEA_MSigDB富集分析:
#包的下载和载入:
devtools::install_github( "ToledoEM/msigdf")
library(msigdf)

共8个类别,我们以C2为例:
#提取C2注释(human):
library(dplyr)
c2<- msigdf.human %>%
filter(category_code == "c2") %>% select(geneset, symbol) %>% as.data.frame
head(c2)
#这里genelist需要的是symbol:
#重新准备genelist文件:
genelist2<- DESeq2$log2FoldChange
names(genelist2) <- rownames(DESeq2)
#genelist过滤(ID转换中丢失的部分基因):
genelist2<- genelist2[names(genelist2) %in% entrez[,1]]
#将genelist按照log2FC值从高到低进行排序:
genelist2<- sort(genelist2,decreasing = T)
head(genelist2)
c2_ges<- GSEA(genelist2,
TERM2GENE= c2,
minGSSize= 10,
maxGSSize= 500,
pvalueCutoff= 0.05,
pAdjustMethod= "BH",
verbose= FALSE,
eps= 0)
c2_ges_result<- c2_ges@result
View(c2_ges_result)

不同数据库的GSEA富集就到这里,可视化方法相同,就不再过多赘述了。
这里针对gene
ID转换,有个小小的tip。当我们使用clusterProfiler或DOSE包进行各类富集分析时,富集结果中的基因为entrez
ID,如果想要在富集结果中以symbol的形式进行直观展示,可以使用setReadable函数进行ID转换。
#在富集结果表中将entrez转换为symbol:
#转换前:
KEGG_ges@result$core_enrichment[1]
#ID转换:
KEGG_ges_set<- setReadable(KEGG_ges,
OrgDb= org.Hs.eg.db,
keyType= "ENTREZID")
#转换后:
KEGG_ges_set@result$core_enrichment[1]
好啦,今天的分享就到这里!