编者按
热图:通过热图可以简单地聚合大量数据,并使用一种渐进的色带来优雅地表现,最终效果一般优于离散点的直接显示,可以很直观地展现空间数据的疏密程度或频率高低。
你还不会画热图吗?
不管您是做蛋白组学、代谢组学或者多层组学研究。热图是文章里常见的图,色彩变化丰富的热图为文章亮色不少。热图可以说是生信中最为常见的图形可视化方式,用以展现表达分布模式。
热图可以用于解决组学研究的什么问题?
根据在蛋白组学、代谢组学、多层组学中多年的研究经验总结。热图一般有两大用途:数据质量控制和展示研究对象的差异变化情况。通过热图,我们能很直观的看到不同分组之间的整体表达模式,因此可以迅速的判断同组之间、各样本间的重复性如何?从而判断实验处理是否正确;数据是否可信可靠符合逻辑。
热图是怎么绘制的呢?
简单的来说,您只要把表达量和组别的数据做成表格,然后调用R语言包就可以绘制热图。这个实际操作起来非常简单快捷易懂。下面跟着小编一起学习一下吧~~
首先安装及加载pheatmap包
#安装包
install.packages("pheatmap")
#加载包
library("pheatmap")
pheatmap包中有很多参数,用于满足我们对热图的调整。虽然有很多参数,通过help(pheatmap),可以查看所有参数的功能。下面给大家讲解一下pheatmap部分参数的实用。
# 数据创建
test = matrix(rnorm(200), 20, 10)
test[1:10, seq(1, 10, 2)] = test[1:10, seq(1, 10, 2)] + 1
test[11:20, seq(2, 10, 2)] = test[11:20, seq(2, 10, 2)] + 2
test[15:20, seq(2, 10, 2)] = test[15:20, seq(2, 10, 2)] + 10
colnames(test) = paste("Test", 1:10, sep = "")
rownames(test) = paste("Gene", 1:20, sep = "")
# 画热图
pheatmap(test,”1.pdf”)
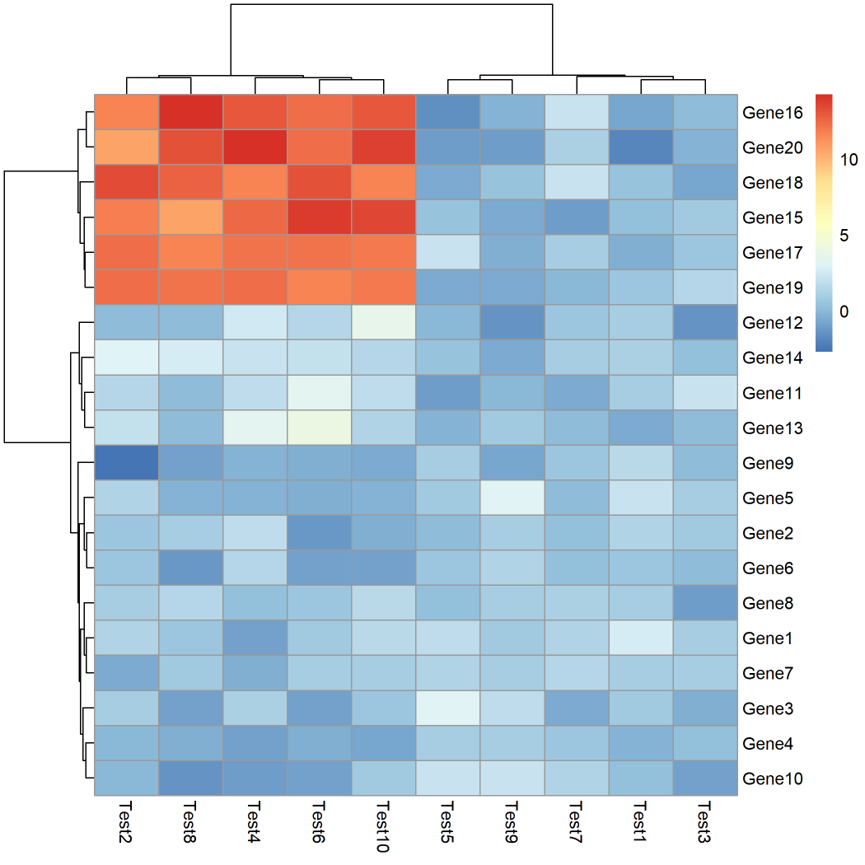
上面的热图很明显不是我们想要的,所以要做一些调整,数据的每一行是基因对应的一个指标,所以对每一行进行数值的中心化和标准化。
pheatmap(test,filename="2.pdf",scale="row")
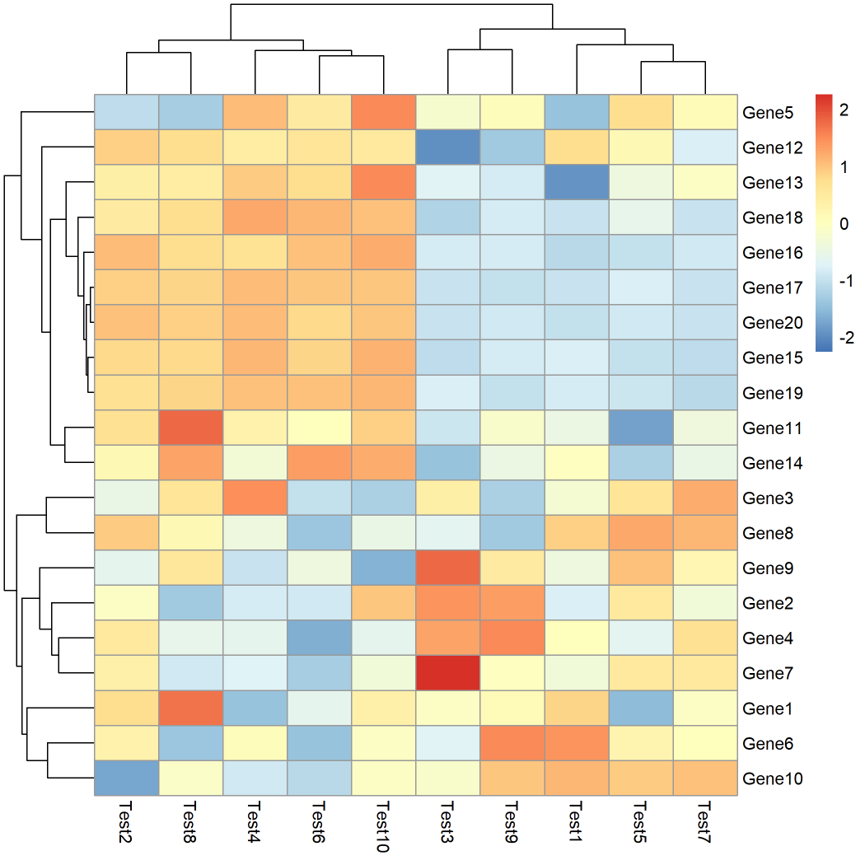
那又该怎么修改颜色呢?下面就给大家展示。
pheatmap(test,filename="3.pdf",scale="row",colour=colorRampPalette(c("blue","white","red"))(100))
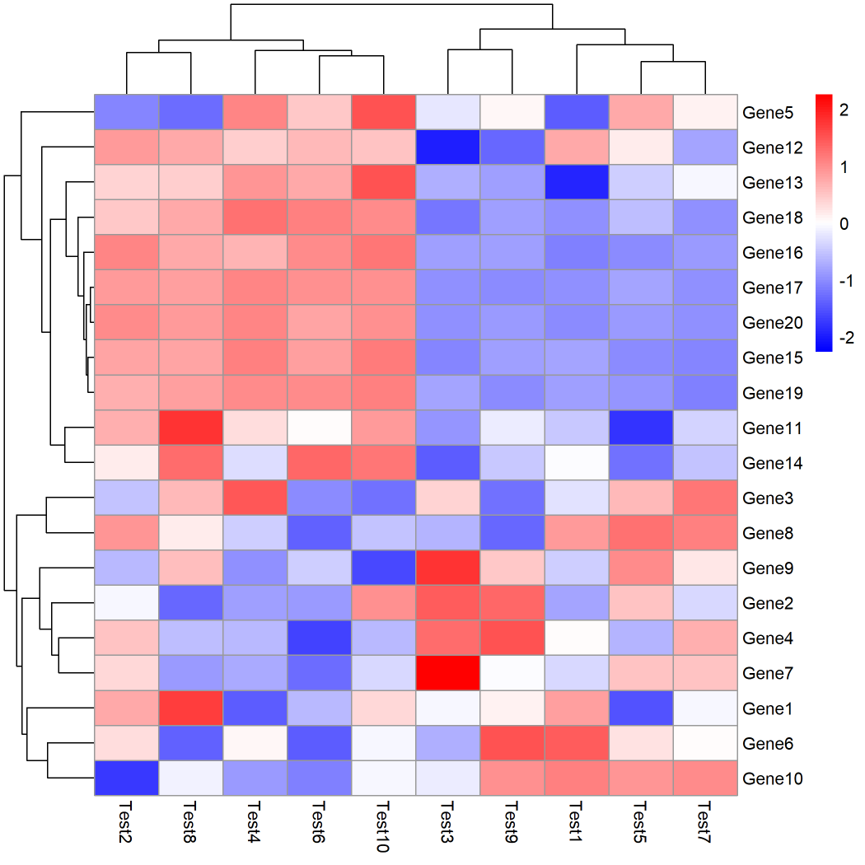
接下来可以对图形进行调整,是否聚类(cluster_rows和cluster_cols);热图单元格大小(cellwidth)和高(cellheight);行聚类树高度(treeheight_row,列聚类是treeheight_col),默认是50。
有感兴趣的小伙伴可以自己尝试一下...
布置作业:
有兴趣的老师可以尝试用上面的语言进行绘制热图哦~~想画更炫热图的老师,可以使用circlize包画一个弯曲的热图。
做蛋白组学、代谢组学、多层组学研究机构,本期针对组学研究做的部分生信分析作图技巧
转自:鹿明生物